



Oligonychus trichardti (Acari: Tetranychidae) associated with Urochloa grasses in Kenya: an integrative taxonomic approach
Espitia-Buitrago, Paula  1
; Perea, Claudia
2
; Odhiambo, Lavender A.
3
; Jauregui, Rosa N.
4
and Chidawanyika, Frank
5
1
; Perea, Claudia
2
; Odhiambo, Lavender A.
3
; Jauregui, Rosa N.
4
and Chidawanyika, Frank
5
1International Centre of Insect Physiology and Ecology (icipe), Nairobi, Kenya & International Center for Tropical Agriculture (CIAT), A.A. 6713, Km 17 recta Cali-Palmira, Palmira, Colombia.
2International Center for Tropical Agriculture (CIAT), A.A. 6713, Km 17 recta Cali-Palmira, Palmira, Colombia.
3International Centre of Insect Physiology and Ecology (icipe), Nairobi, Kenya.
4International Center for Tropical Agriculture (CIAT), A.A. 6713, Km 17 recta Cali-Palmira, Palmira, Colombia.
5✉ International Centre of Insect Physiology and Ecology (icipe), Nairobi, Kenya & Department of Zoology and Entomology, University of the Free State, P.O. Box 339, Bloemfontein 9300, South Africa.
2026 - Volume: 66 Issue: 2 pages: 469-481
https://doi.org/10.24349/swpy-fpiiOriginal research
Keywords
Abstract
Introduction
The livestock sector is central to Africa's economy, with cattle contributing about 40% of the continent's agricultural GDP and nearly one-third of the global cattle population (Florez et al. 2024). In East African countries, livestock supports food security, income generation, and employment, contributing to local economies and sustaining the livelihoods of millions (East African Community 2022). To close the 40% feed gap in the region, improved forages such as Brachiaria cultivars (Urochloa P.Beauv. (syn. Brachiaria (Trin.) Griseb). have been shown to increase feed availability and livestock productivity, including milk yields (González et al. 2016; Burkart and Mwendia 2024). Apart from their role as forages, these grasses play a critical role in climate change mitigation due to their extensive root system that promotes soil organic carbon sequestration and biological nitrification, and low-emission livestock systems (Baptistella et al. 2020). However, their adoption remains low, in part due to high seed costs and agronomic challenges such as pest and disease pressure (Junca Paredes et al. 2023). Among these constraints, spider mite infestations have emerged as a significant limitation for Urochloa in Kenya, leading farmers to prefer cultivars with both drought and mite tolerance (Cheruiyot et al. 2020).
To the best of our knowledge, there are no control strategies with efficacy against this threat(Cheruiyot et al. 2018; Mutisya et al. 2018). Several spider mite species (Acari: Tetranychidae) have been documented infesting Urochloa grasses. These include Oligonychus trichardti Meyer, 1974, reported by Cheruiyot et al. (2018), and Tetranychus urticae Koch, 1836, reported by Mutisya et al. (2018). Furthermore, according to the Spider Mites Web database (Migeon and Dorkeld 2026), O. grypus (Baker & Pritchard, 1960) and O. indicus (Hirst, 1923) are also recorded as infesting this host within the region. The genus Oligonychus Berlese (Acari: Tetranychidae) is the most diverse in the family Tetranychidae, with over 200 described species that exhibit a wide range of biological traits, life cycles, and feeding habits (Mushtaq et al. 2022). Accurate species identification is therefore a crucial step towards development of effective management strategies, as mite species differ in the type and severity of plant damage they cause. This is particularly important for host-plant resistance, a first line of plant defense against herbivory, since the genetic and physiological responses of the host plant can be highly specific to the attacking mite species. In other Poaceae species including maize (Zea mays L.), resistance mechanisms have been shown to differ depending on the infesting species such as the highly polyphagous generalist T. urticae and the less polyphagous with a preference for grasses Oligonychus pratensis (Bui et al. 2018, 2021; Barnes et al. 2024). Such species-specific comparative studies of herbivory impacts on target plants are only possible following accurate taxonomic species identification.
However, accurate species identification remains challenging due to the requirement to collect adults, especially males, and an unwillingness to engage with specialist acarologists (Ovalle et al. 2020; İnak et al. 2022). Within the genus Oligonychus, traditional taxonomy has relied on the morphology of the male genitalia, particularly the aedeagus, which requires high skill and precise lateral mounting for accurate observation, making it prone to erroneous classifications. To improve the classification Mushtaq et al. (2024) recently proposed an updated taxonomic key based on additional morphological traits including female and male traits that classify species into subgenera, species groups, and subgroups. In parallel, modern taxonomy increasingly integrates morphological analysis with molecular barcoding to improve the accuracy and robustness of species identification (Razuvaeva et al. 2023; Lu et al. 2024). This integrative approach has been successfully applied within Oligonychus, with (Mushtaq et al. 2023b) confirming the identity of 42 species and clarifying phylogenetic relationships within the genus.
Materials and methods
Mite colony and collection
In February 2024, a rearing stock of 80 adult spider mites was collected from a symptomatic plot of an interspecific Urochloa hybrid cv. Cayman at a livestock farm in Mbita Point, Kenya (0°25′59.7''S, 34°12′18.4''E). The infested plants showed characteristic symptoms of spider mite infestation: foliar chlorosis and necrotic spots (Espitia-Buitrago et al. 2025). The mites with the same morphotype under the stereoscope were transferred to clean uninfested plants of interspecific Urochloa hybrid cv. Mulato II for colony establishment. Two colonies were maintained, each within a cage containing two host plants, and kept in a shaded area under natural environmental conditions. Host plants were replaced every two weeks with fresh, uninfested plants to sustain the colonies. In May 2024, a total of 70 individuals including adult females, males, and nymphs, were collected and preserved in 70% ethanol for subsequent slide mounting or DNA extraction.
Morphological and taxonomical identification
A total of 46 individuals were slide-mounted using Hoyer's medium following the protocol by (Krantz and Walter 2009). In brief, one to two drops of Hoyer's mounting medium were placed on a microscope slide, and individual mites were carefully transferred using a fine Camel hairbrush and needle. Each specimen was gently immersed in the medium, and both dorsal and ventral positions were examined for overall evaluation of diagnostic features. A circular cover slide was placed slowly whilst avoiding trapping of air bubbles. The mounted slides were dried in an oven at 45 °C for 3–5 days, with regular checks to prevent overheating or bubble formation. Once fully dried, the edges of the cover slips were sealed to preserve the mounts.
Mounted specimens were examined under a NIKON Eclipse Ci microscope equipped with a NIKON Sight D5 FI2 digital camera. Taxonomic identification was carried out using morphological keys proposed by Bolland et al. (1998); the species group, and subgroups species followed the criteria established by Mushtaq et al. 2021a, b, 2023b, 2024. The terminology, abbreviations and leg chaetotaxy used in the manuscript follows those of Lindquist 1985.
DNA extraction and PCR amplification
Molecular analyses were conducted on individuals from the colonies to provide diagnostic markers. Thus, genomic DNA was extracted from ten individual adult females, with each extraction performed on a single specimen. Females were selected due to their high prevalence in the colonies and their larger body size, which ensures a more reliable DNA yield for PCR amplification. The extraction followed a protocol adapted from Landry et al. (1993), as previously reported by Espitia-Buitrago et al. (2024). Briefly, each specimen was homogenized in 160 μL of lysis buffer (200 mM Tris-HCl pH 8.0, 70 mM EDTA pH 8.0, 2 M NaCl, and 20 mM sodium metabisulfite) using a sterile plastic pestle. After homogenization, 40 μL of 5% SDS was added to each tube, mixed by inversion, and incubated at 55 °C for 2 hours to facilitate cell lysis.
Following incubation, samples were centrifuged at 14,000 g for 15 minutes at room temperature. The supernatant was transferred to fresh tubes and mixed with 90 μL of 10 M ammonium acetate and 200 μL of cold isopropanol. Samples were then stored at –20 °C for 2 hours to allow DNA precipitation. DNA pellets were recovered by centrifugation (14,000 g, 15 min), washed with 500 μL of ethanol 70%, and air-dried. Finally, DNA was re-suspended in 30 μL of double-distilled water and stored at –20 °C. The integrity of the extracted genomic DNA was confirmed by electrophoresis on a 2% agarose gel (2 μL per sample).
The mitochondrial cytochrome c oxidase subunit I (COI) and nuclear internal transcribed spacer 2 (ITS2) regions were amplified from DNA extracts using PCR primers previously described by Navajas et al. (1996) for COI and Ben-David et al. (2007) for ITS2. These primer sets were selected to ensure compatibility with existing Oligonychus GenBank data and have been successfully employed in recent Oligonychus molecular studies (Ben-David et al., 2007; Mushtaq et al., 2023:COI-forward (5′-TGATTTTTTGGTCACCCAGAAG-3′) and COI-reverse (5′-TACAGCTCCTATAGATAAAAAC-3′); ITS2-forward (5′-GTCACATCTGTCTGAGAGTTGAGA-3′) and ITS2-reverse (5′-GTARCCTCACCTRMTCTGAGATC-3′).
PCR reactions were performed in 25 µL volumes containing 2.5 µL of 10× PCR buffer, 2.0 mM MgCl₂, 0.2 mM dNTPs, 0.2 µM of each primer, 1 U Taq DNA polymerase, and 2 µL template DNA. Thermal cycling conditions were: (i) initial denaturation at 94 °C for 3 min; (ii) 35 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C (COI) or 54 °C (ITS2) for 30 s, and extension at 72 °C for 1 min; and (iii) final extension at 72 °C for 5 min.
PCR products were visualized on 1.5% agarose gels stained with SYBR Safe. Successfully amplified products were sent to the International Livestock Research Institute (ILRI) in Nairobi, Kenya, for Sanger sequencing.
Bioinformatic analysis
Forward and reverse chromatograms were assembled into consensus sequences in Geneious Prime v.2024.0.1 (https://www.geneious.com ![]() ). Low-quality ends (Phred score >20) and ambiguous bases were trimmed manually. The resulting sequences were quality-checked in MEGA v.12 (Stecher and Kumar, 2025). As no nucleotic variation was detected among the 10 sequenced individuals for either marker, a single representative consensus sequence per gene (COI and ITS2) was retained for downstream analyses.
). Low-quality ends (Phred score >20) and ambiguous bases were trimmed manually. The resulting sequences were quality-checked in MEGA v.12 (Stecher and Kumar, 2025). As no nucleotic variation was detected among the 10 sequenced individuals for either marker, a single representative consensus sequence per gene (COI and ITS2) was retained for downstream analyses.
Representative sequences were compared against the NCBI GenBank database using BLASTn (Altschup et al. 1990) to identify closest related taxa and guide selection of comparative sequences. Additional sequences of Oligonychus spp. were retrieved from GenBank (Clark et al., 2016) to maximize species representation (Table S1). Sequences were selected based on (i) sufficient overlap with the amplified region, (ii) sequence length and quality, and (iii) reliable taxonomic annotation. Sequences with short alignment overlaps, excessive ambiguities or questionable identification were excluded. To reduce redundancy, a maximum of two accessions per species were retained when available, prioritizing sequences from independent submissions or different geographic origins.
Multiple sequence alignment was performed using MAFFT (default parameters), followed by alignment refinement using MUSCLE implemented in MEGA V.12. Alignments were inspected manually and trimmed to a common overlapping region prior to phylogenetic inference. For COI sequences, alignments were translated into amino acids using the invertebrate mitochondrial genetic code to confirm the absence of internal stop codons and frameshifts. Additional manual trimming of the 5′ region was performed when necessary to correct the reading frame.
Phylogenetic reconstruction was conducted in MEGA V.12 using the Maximum Likelihood (ML) method with 1,000 bootstrap replicates. The best-fit nucleotide substitution models were selected based on the lowest Bayesian Information Criterion (BIC) score as implemented in MEGA. Gaps and missing data were treated using partial deletion with 80% site coverage cutoff. For the COI dataset, Tetranychus urticae and Amphitetranychus viennensis were included as external taxa to root the tree. For the ITS2 dataset, Tetranychus sp. (KT361605.1) was used as outgroup. Clades with bootstrap support values >60% were collapsed for visualization
Phylogenetic analyses were conducted primarily to assess species-level clustering and diagnostic placement rather than to infer deep evolutionary relationships within the genus.
The GeneBank accession numbers included in the phylogenetic analysis are provided in Supplementary Table S1.
The newly generated sequences from this study were deposited in GeneBank under accession numbers PX133189 and PX131128.
Results
Morphological identification
A total of 36 females and three males were identified. All specimens presented the same morphotype identified as Oligonychus (Reckiella) trichardti Meyer, 1974. All observed morphological characters corresponded to descriptions in (Reck 1959; Meyer 1974, 1987) and subsequent diagnostics by (Mushtaq et al. 2021a, 2023a, b).
Family Tetranychidae Donnadieu
Subfamily Tetranychinae Berlese
Tribe Tetranychini Reck
Genus Oligonychus (Berlese 1886)
Oligonychus Berlese, 1886: 24** Following (Mushtaq et al. 2021a).
Type species — Heteronychus brevipodus (Berlese & Targioni Tozzetti 1878): 255
Opisthosoma with 11 pairs of dorsal setae (c1-3, d1-2, e1-2, f1-2, h2-3; setae h2 and h3 usually inserted ventrally); clunal setae h1 always absent (Fig. 1A). Empodia well developed, claw-like with proximoventral hairs (except male leg I with hairs modified into spur) that are as long as or shorter than empodial claw on most of legs (Fig. 1C); Two pairs of duplex setae on tarsus I, distal and adjacent (Fig. 2C).
Subgenus Reckiella (Tuttle & Baker 1968)
Diagnosis (based on male) — Following to (Mushtaq et al. 2021a). Male aedeagus with shaft bending dorsad, or shaft initially bends dorsad then distal part turned ventrad, upturned part usually without tapering end, distally forming knob, sigmoidal shape and blunt or rounded tip. (Fig. 3)
exsiccator species group
gossypii species subgroup
Diagnosis (based on female) — Following to (Mushtaq et al. 2021a). More than seven (eight, nine or rarely ten) tactile setae on tibia I (Fig. 2B), and dorsal hysterosoma with various patterns of striae: longitudinal, irregular longitudinal, oblique, and with/without forming clear/inverted V/U-shaped striae between both setal pairs e1-e1 and f1-f1, or posterior to f1-f1, and/or striae forming a ''hourglass-shaped″between e1-f2 area (Fig. 2A). This subgroup includes ten species, between them O. trichardti.
Oligonychus (Reckiella) trichardti Meyer, 1974
Oligonychus (Reckiella) trichardti Meyer, 1974): 262
Figures 1-3
Host plant type — Poaceae sp. Meyer, 1974. Type Distribution: South Africa and Kenya
Host plant records — Brachiaria brizantha (Meyer 1987); Brachiaria sp. (Cheruiyot et al. 2018); Heteropogon contortus; Hyparrhenia filipendula (Meyer 1987); Hyperthelia dissoluta (Meyer 1987).

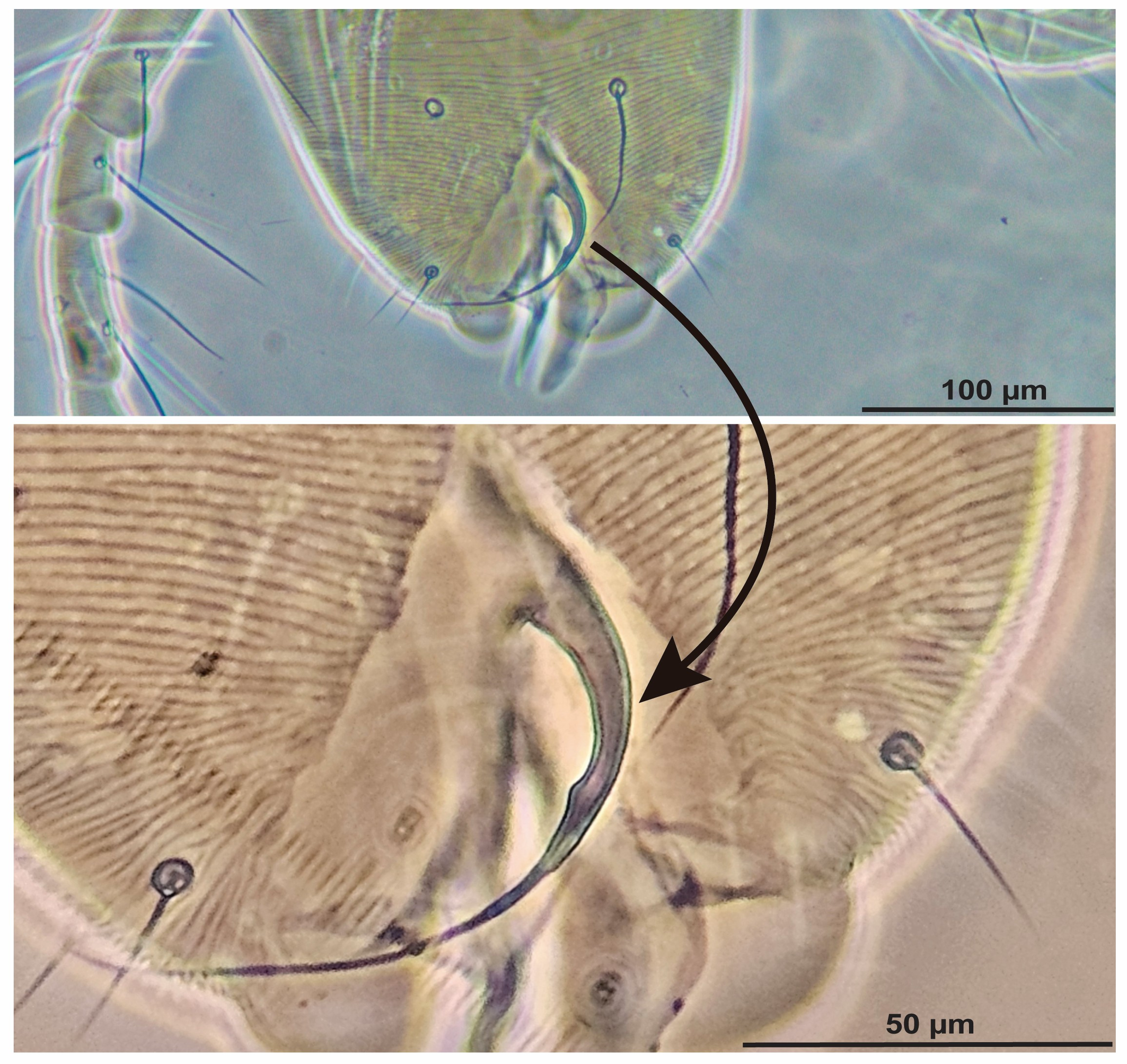
Diagnosis (based on males) — The short shaft of the aedeagus curves dorsad, forming a knob which is provided with a small, acute anterior projection, while the posterior projection drawn out into a very long and slender stylet that is directed anterodorsally (Fig. 3); Gnathosoma. The palptarsus bears a terminal sensillum (spineret) that is slightly more than twice as long as broad (Fig. 1F).
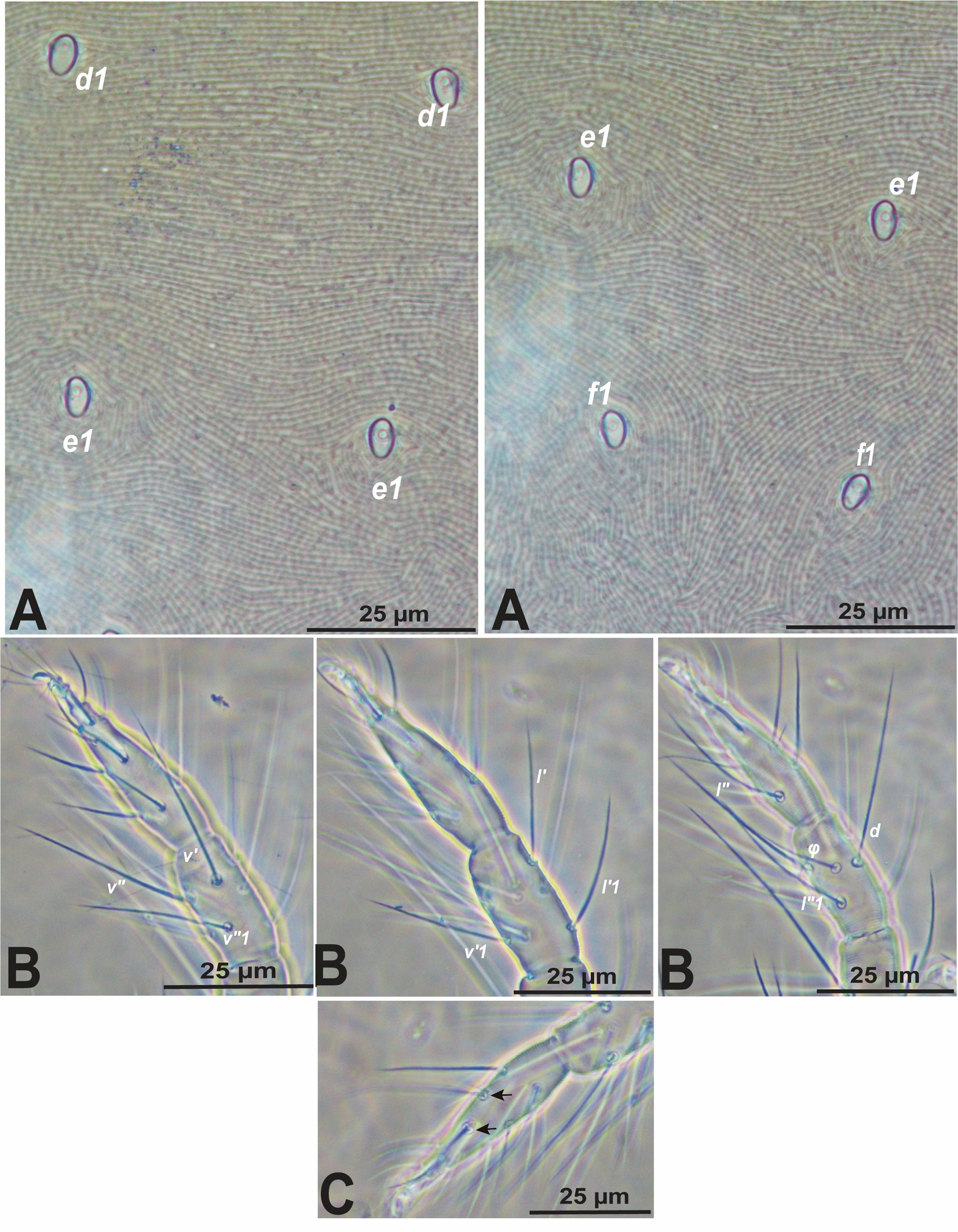
Diagnosis (based on females) — Hysterosoma with longitudinal or irregularly longitudinal striae between fourth pair of dorsocentral setae (Fig. 2A). The palptarsus bears a terminal sensillum (spineret) one and a half times as long as broad (Fig. 1E); peritreme terminates in a simple bulb (Fig. 1B).
Phylogenetic analysis based on COI gene sequences
The COI alignment comprised 17 nucleotide sequences of 408 bp after manual trimming to a common overlapping region and adjusting the reading frame. . Phylogenetic reconstruction was performed using the Maximum Likelihood method under the General Time Reversible (GTR) model with gamma-distributed rate variation among sites (61.96%). The tree with the highest log likelihood (-1,973.07) is shown in Figure 4. The COI alignment used for phylogenetic analysis is provided in Supplementary File S2.
The Maximum Likelihood (ML) analysis of COI sequences focused on Oligonychus (subgenus Reckiella) and included Tetranychus urticae (DQ017588.1) and Amphitetranychus viennensis (FJ997620.1) as external taxa to root the tree (Fig. 4). Several species represented by two accessions formed strongly supported clades, including O. orthius (BS = 99), O. rubicundus (BS = 100), O. modestus (BS = 100), O. afrasiaticus (BS = 100), and O. biharensis (BS = 100), demonstrating clear species-level clustering in the COI dataset.
The studied specimen (Oligonychus trichardti PX133189.1) was placed within the Reckiella clade and recovered as sister to the O. afrasiaticus species, with moderate bootstrap support (BS = 72). Deeper relationships among several Reckiella species were weakly to moderately supported, indicating limited resolution among closely related taxa based on this single mitochondrial marker. Overall, the COI tree supports placement of the Kenyan specimen within Oligonychus (Reckiella) and its distinction from other sampled Reckiella species.

Phylogenetic analysis based on ITS2 gene sequences
The ITS2 alignment comprised 13 nucleotide sequences and 601 bp after manual trimming to a common overlapping region. . Phylogenetic reconstruction was performed using the Maximum Likelihood method under the General Time Reversible (GTR) model with gamma-distributed rate variation among sites (G = 2.4535), applying partial deletion (80% site coverage) which resulted in a final length of 429bp, and a proportion of invariant sites (51.48%). The tree with the highest log likelihood (−1,823.41) is shown in Figure 5. The ITS2 alignment used for phylogenetic analysis is provided in Supplementary File S2.
Species represented by multiple accessions formed strongly supported clades, including O. tylus (BS = 100), O. afrasiaticus (BS = 100), O. washingtoniae (BS = 100), and O. indicus (BS = 100), indicating clear species-level resolution using ITS2.
The studied specimen (O. trichardti PX131128.1) was placed within the Reckiella assemblage and grouped near O. washingtoniae, with moderate support at deeper nodes (BS = 60–64). As in the COI analysis, backbone relationships among closely related Reckiella species received moderate bootstrap support, suggesting limited resolution of interspecific relationships based on this single nuclear marker.

Discussion
This study presents the first integrative taxonomic identification of Oligonychus trichardti from Kenya, combining morphological examination with newly generated COI and ITS2 sequence data. The morphological characters observed in our specimens correspond precisely with the original description of Meyer (1974), confirming the presence of this species on Urochloa grasses in East Africa. Particularly, the aedeagus morphology is a crucial diagnostic feature for this species with a configuration that distinguishes O. trichardti from other members of the gossypii species subgroup. Our observations confirm that the Kenyan specimens align with the ''gossypii-type'' morphology.
Integrative taxonomic approach and species identification
The sequences presented here should be regarded as the first diagnostic references for the species.
Morphological examination, particularly of the male aedeagus, dorsal striation pattern, and leg chaetotaxy, provided the basis for species identification. The molecular data do not replace morphology but complement it by supplying reference sequences that can assist in future identifications, especially when males are unavailable or when only immature stages are collected. In this context, the integrative approach strengthens diagnostic reliability without extending beyond the limits of the data.
Accurate species identification is essential for understanding host associations and developing targeted management strategies in forage systems. The confirmation of O. trichardti on Urochloa in Kenya expands the documented molecular reference framework for this species and facilitates future diagnostic efforts in East African agroecosystems.
Phylogenetic placement within Reckiella
Both COI and ITS2 analyses placed the Kenyan specimen within the subgenus Reckiella, consistent with its morphological assignment based on aedeagal orientation. Species represented by multiple accessions formed strongly supported clades in both datasets, demonstrating that the markers employed provide effective species-level discrimination within the sampled taxa.
The placement of O. trichardti as sister to the O. afrasiaticus clade in the COI tree received moderate bootstrap support, while ITS2 placed the species within the broader Reckiella assemblage with limited resolution among closely related taxa. These results indicate that single-locus datasets, particularly short mitochondrial fragments, provide reliable species-level clustering but limited resolution of deeper interspecific relationships within the genus.
Given the restricted taxon sampling and fragment length, no conclusions are drawn here regarding evolutionary relationships among species subgroups within Reckiella. Rather, the molecular analyses are interpreted conservatively as supporting the placement of the Kenyan specimens within Oligonychus (Reckiella) and confirming their distinction from other sampled species.
Biogeographic significance
This study confirms the documented geographic range of O. trichardti in East Africa (Cheruiyot et al., 2018), previously known primarily endemic to South Africa where it was originally described (Meyer, 1974). The species has been reported from various Poaceae hosts across Africa (Meyer 1987; Cheruiyot et al. 2018), but the generation of molecular data confirmation from Kenya populations represents an important diagnostic contribution. The identification of O. trichardti on Urochloa grasses in Kenya is consistent with its known host range and ecological preferences for gramineous plants.
Implications for forage crop management
The accurate identification of spider mite species is crucial for developing effective integrated pest management strategies. Our confirmation of O. trichardti as a pest of Urochloa grasses in Kenya supports previous reports (Cheruiyot et al. 2018) and provides molecular evidence for diagnostic applications and management planning. The species-specific identification is particularly important given that closely related Oligonychus species may differ in their biology, seasonal patterns, and susceptibility to control measures.
The integration of Urochloa cultivars into East African livestock systems represents a significant opportunity to address feed gaps and improve productivity. However, spider mite pressure has been identified as a constraint to adoption (Cheruiyot et al. 2022). Establishing the identity of the specific mite species involved is the first step in developing targeted management approaches, including the identification of resistant cultivars and appropriate miticide selection.
Future research directions
This study provides diagnostical molecular references for O. trichardti but does not attempt to resolve deeper phylogenetic relationships within the genus. Broader taxon sampling and additional molecular markers will be necessary to clarify interspecific relationships within the subgenus Reckiella.
Expanded geographic sampling of O. trichardti populations across Africa would allow evaluation of genetic variability and population structure, which remain unexplored. In addition, ecological studies examining seasonal dynamics, host plant interactions, and damage thresholds on Urochloa grasses would support the development of sustainable management strategies.
The integrative taxonomic framework applied here demonstrates the value of combining morphology with molecular data for reliable species identification in taxonomically complex mite groups. By providing verified reference sequences linked to morphologically identified specimens, this study contributes to improving diagnostic capacity and supporting applied research in forage production systems.
Acknowledgements
We are grateful to Edwin Quintero-Rodriguez for his expertise in the taxonomic identification of this species and his valuable contributions to the morphological section of this study. We also thank Adrian Kimani for his help in the sampling and collection of the specimens, and the Mbita farmers who granted us permission for sampling, and the icipe Plant Health Theme and Tropical Forages Team at CIAT for their valuable contributions. Additionally, we would like to thank the editor and the reviewers for their constructive comments and suggestions, which helped to improve the quality of this manuscript.
This work was supported by the CGIAR Breeding for Tomorrow Science Program and the CGIAR Accelerated Breeding Initiative. CGIAR research is supported by contributions to the CGIAR Trust Fund. CGIAR is a global research partnership for a food-secure future dedicated to transforming food, land, and water systems in a climate crisis. The authors also gratefully acknowledge the financial support to icipe by the EU DG INTPA through the RMRN-EA (grant number: NDICI AFRICA/2024/458-631), Swedish International Development Cooperation Agency (Sida); the Swiss Agency for Development and Cooperation (SDC); the Australian Centre for International Agricultural Research (ACIAR); the Government of Norway; the German Federal Ministry for Economic Cooperation and Development (BMZ); and the Government of the Republic of Kenya. The views expressed herein do not necessarily reflect the official opinion of the donors.
References
- Altschup S.-F., Gish W., Miller W., Myers E.-W., Lipman D.-J. 1990. Basic Local Alignment Search Tool. J. Mol. Biol., 215: 403-410. https://doi.org/10.1016/S0022-2836(05)80360-2
- Baptistella J.-L.-C., de Andrade S.-A.-L., Favarin J.-L., Mazzafera P. 2020. Urochloa in Tropical Agroecosystems. Front. Sustain. Food Syst., 4: 119. https://doi.org/10.3389/fsufs.2020.00119
- Barnes C.-L., Wickwar D., Yost M., Spears L.-R., Ramirez R.-A. 2024. The effects of water-stress, temperature, and plant traits on the outbreak potential of a specialist and generalist spider mite species (Acari: Tetranychidae). J. Appl. Entomol., 148: 13-25. https://doi.org/10.1111/jen.13204
- Ben-David T., Melamed S., Gerson U., Morin S. 2007. ITS2 sequences as barcodes for identifying and analyzing spider mites (Acari: Tetranychidae). Exp. Appl. Acarol., 41: 169-181. https://doi.org/10.1007/s10493-007-9058-1
- Berlese A. 1886. Acari dannosi alle piante coltivate. Padova: Sacchetto. pp. 31.
- Berlese A., Targioni Tozzetti A. 1878. Relazione interno ai lavori della Stazione di entomologia agraria di Firenze per l′anno 1876. Acaridei Annali di Agricoltura, 1: 242-275.
- Bolland H.-R., Gutierrez J., Flechtmann C.-H.-W. 1998. World catalogue of the spider mite family (Acari: Tetranychidae). Leiden: Brill. pp. 392. https://doi.org/10.1163/9789004630536
- Bui H., Greenhalgh R., Gill G.-S., Ruckert A., Enslin S., Clark R.-M. 2021. Maize Inbred Line B96 Is the Source of Large-Effect Loci for Resistance to Generalist but Not Specialist Spider Mites. Front. Plant Sci., 12: 693088. https://doi.org/10.3389/fpls.2021.693088
- Bui H., Greenhalgh R., Ruckert A., Gill G.-S., Clark R.-M. 2018. Generalist and specialist mite herbivores induce similar defense responses in maize and barley but differ in susceptibility to benzoxazinoids. Front. Plant Sci., 9: 1222. https://doi.org/10.3389/fpls.2018.01222
- Burkart S., Mwendia S. 2024. Forage Seed Systems to Close the Ruminant Feed Deficit in Eastern Africa. Grasses, 3: 333-354. https://doi.org/10.3390/grasses3040025
- Cheruiyot D., Chidawanyika F., Midega C.-A.-O., Pittchar J.-O., Pickett J.-A., Khan Z.-R. 2022. Field evaluation of a new third generation push-pull technology for control of striga weed, stemborers, and fall armyworm in western Kenya. Exp. Agric., 58: e4. https://doi.org/10.1017/S0014479721000260
- Cheruiyot D., Midega C.-A.-O., Pittchar J.-O., Pickett J.-A., Khan Z.-R. 2020. Farmers' perception and evaluation of brachiaria grass (Brachiaria spp.) genotypes for smallholder cereal-livestock production in East Africa. Agriculture, 10: 268. https://doi.org/10.3390/agriculture10070268
- Cheruiyot D., Midega C.-A.-O., Ueckermann E.-A., Van den Berg J., Pickett J.-A., Khan Z.-R. 2018. Genotypic response of brachiaria (Urochloa spp.) to spider mite (Oligonychus trichardti) (Acari: Tetranychidae) and adaptability to different environments. Field Crops Res., 225: 163-169. https://doi.org/10.1016/j.fcr.2018.06.011
- East African Community. 2022. Livestock development. Agriculture & Food Security [Internet]. [Consultado 14 Ene 2026]. Available from: https://www.eac.int/agriculture/livestock-development/
- Espitia-Buitrago P., Cotes-Torres J.-M., Hernández L.-M., Chidawanyika F., Jauregui R. 2025. Enhancing Phenotyping Accuracy for Selection of Urochloa spp. Tolerant Genotypes to Red Spider Mite (Oligonychus trichardti). Grass Forage Sci., 80. https://doi.org/10.1111/gfs.70007
- Espitia-Buitrago P., Perea C., Chidawanyika F., Jauregui R. 2024. Urochloa red spider mite (Acari: Tetranychidae) identification: morphological and molecular techniques [Report]. Cali: Alliance of Bioversity International and CIAT.
- Florez J.-F., Karimi P., Paredes J.-J.-J., Birnholz C., Junca-Paredes J.-J., Schiek B., Peters M., Notenbaert A. 2024. Developments, bottlenecks, and opportunities in seed markets for improved forages in East Africa: The case of Kenya. Grassl. Res., 3: 79-96. https://doi.org/10.1002/glr2.12073
- González C., Schiek B., Mwendia S., Prager S. 2016. Improved forages and milk production in East Africa [Report]. Cali: CIAT. No. 421.
- İnak E., Çobanoğlu S., Auger P., Migeon A. 2022. Molecular identification and phylogenetic analysis of spider mites (Prostigmata: Tetranychidae) of Turkey. Exp. Appl. Acarol., 87: 195-205. https://doi.org/10.1007/s10493-022-00728-5
- Junca-Paredes J.-J., Florez J.-F., Enciso-Valencia K.-J., Karimi P., Burkart S. 2023. Potential Forage Hybrid Markets for Enhancing Sustainability and Food Security in East Africa. Foods, 12: 1607. https://doi.org/10.3390/foods12081607
- Krantz G.-W., Walter D.-E. 2009. A manual of acarology, 3rd edn. Lubbock: Texas Tech University Press. pp. 707.
- Landry B.-S., Dextraze L., Boivin G. 1993. Random amplified polymorphic DNA markers for DNA fingerprinting and genetic variability assessment of minute parasitic wasp species (Hymenoptera: Mymaridae and Trichogrammatidae) used in biological control programs of phytophagous insects. Genome, 36: 580-587. https://doi.org/10.1139/g93-078
- Lindquist E.-E. 1985. External anatomy and notation of structures. In: Helle W., Sabelis M.-W. (Eds). Spider mites: their biology, natural enemies, and control. Amsterdam: Elsevier. p. 3-28.
- Lu C., Hao S.-D., Ha P.-Z., Song Z.-S., Sun J.-T., Xue X.-F., Hong X.-Y. 2024. A multiplex direct PCR method for the rapid and accurate discrimination of three species of spider mites (Acari: Tetranychidae) in fruit orchards in Beijing. Exp. Appl. Acarol., 92: 403-421. https://doi.org/10.1007/s10493-023-00900-5
- Meyer M.-K.-P. 1974. A revision of the Tetranychidae of Africa (Acari) with a key to the genera of the world. Entomology Mem. Dep. Agric. Tech. Serv. Repub. S. Afr., 36: 1-291.
- Meyer M.-K.-P. 1987. African Tetranychidae (Acari: Prostigmata) with reference to the world genera. Entomology Mem. Dep. Agr. Wat. Supply Repub. S. Afr., 69: 1-175.
- Migeon A., Dorkeld F. Spider Mites Web: a comprehensive database for the Tetranychidae [Internet]. [14 Jan 2026]. Montpellier: INRAE/CBGP. Available from: https://www1.montpellier.inrae.fr/CBGP/spmweb/
- Mushtaq H.-M.-S., Alatawi F.-J., Kamran M., Flechtmann C.-H.-W. 2021a. The genus Oligonychus Berlese (Acari, Prostigmata, Tetranychidae): taxonomic assessment and a key to subgenera, species groups, and subgroups. ZooKeys, 1079: 89-127. https://doi.org/10.3897/zookeys.1079.75175
- Mushtaq H.-M.-S., Kamran M., Alatawi F.-J. 2021b. Two new life types and assessment of web-associated behavioral characteristics of some Oligonychus species on various host plants. Exp. Appl. Acarol., 83: 211-227. https://doi.org/10.1007/s10493-020-00579-y
- Mushtaq H.-M.-S., Kamran M., Alatawi F.-J. 2022. New species, new records, and re-descriptions of two species of the genus Oligonychus Berlese (Acari: Prostigmata: Tetranychidae) from Saudi Arabia. Syst. Appl. Acarol., 27: 2568-2582. https://doi.org/10.11158/saa.27.12.9
- Mushtaq H.-M.-S., Kamran M., Alatawi F.-J. 2024. The subgenus Oligonychus (Oligonychus) Berlese (Acari, Prostigmata, Tetranychidae), diagnostic keys to world species, and taxonomic notes. Eur. Zool. J., 91: 559-567. https://doi.org/10.1080/24750263.2024.2350197
- Mushtaq H.-M.-S., Kamran M., Saleh A.-A., Alatawi F.-J. 2023a. Evidence for Reconsidering the Taxonomic Status of Closely Related Oligonychus Species in punicae Complex (Acari: Prostigmata: Tetranychidae). Insects, 14: 3. https://doi.org/10.3390/insects14010003
- Mushtaq H.-M.-S., Saleh A.-A., Kamran M., Alatawi F.-J. 2023b. Molecular-Based Taxonomic Inferences of Some Spider Mite Species of the Genus Oligonychus Berlese (Acari, Prostigmata, Tetranychidae). Insects, 14: 192. https://doi.org/10.3390/insects14020192
- Navajas M., Gutierrez J., Lagnel J., Boursot P. 1996. Mitochondrial cytochrome oxidase I in tetranychid mites: a comparison between molecular phylogeny and changes of morphological and life history traits. Mol. Biol. Evol., 13: 422-427. https://doi.org/10.1093/oxfordjournals.molbev.a025604
- Ovalle T.-M., Vásquez-Ordóñez A.-A., Jimenez J., Moreno M.-C., Hernandez L.-M., Wyckhuys K.-A.-G., Lopez-Lavalle L.-A.-B. 2020. A simple PCR-based method for the rapid and accurate identification of spider mites (Tetranychidae) on cassava. Sci. Rep., 10: 18361. https://doi.org/10.1038/s41598-020-75743-w
- Razuvaeva A.-V., Ulyanova E.-G., Skolotneva E.-S., Andreeva I.-V. 2023. Species identification of spider mites (Tetranychidae: Tetranychinae): a review of methods. Vavilovskii Zhurnal Genet. Selektsii, 27: 240-249. https://doi.org/10.18699/VJGB-23-30
- Reck G.-F. 1959. A key to the tetranychoid mites. Tbilissi: Akademii Nauk Gruzinskoi SSR. pp. 152.
- Seeman O.-D., Beard J.-J. 2011. Identification of exotic pest and Australian native and naturalised species of Tetranychus (Acari: Tetranychidae). Zootaxa, 2976: 1-72. https://doi.org/10.11646/zootaxa.2976.1.1
- Stecher G., Kumar S. 2021. MEGA12: Molecular Evolutionary Genetics Analysis Version 12. Mol. Biol. Evol., 38: 3022-3027. https://doi.org/10.1093/molbev/msab120
- Tuttle D.-M., Baker E.-W. 1968. Spider mites of Southwestern United States and a revision of the family Tetranychidae. Tucson: University of Arizona Press. pp. 143.



2025-10-23
Date accepted:
2026-05-12
Date published:
2026-05-18
Edited by:
Migeon, Alain

This work is licensed under a Creative Commons Attribution 4.0 International License
2026 Espitia-Buitrago, Paula; Perea, Claudia; Odhiambo, Lavender A.; Jauregui, Rosa N. and Chidawanyika, Frank
 Download article
Download articleDownload the citation
RIS with abstract
(Zotero, Endnote, Reference Manager, ProCite, RefWorks, Mendeley)
RIS without abstract
BIB
(Zotero, BibTeX)
TXT
(PubMed, Txt)