



Molecular evolutionary trends and biosynthesis pathways in the Oribatida revealed by the genome of Archegozetes longisetosus
Brückner, Adrian  1
; Barnett, Austen A.
2
; Bhat, Prashant
3
; Antoshechkin, Igor A.
4
and Kitchen, Sheila A.
5
1
; Barnett, Austen A.
2
; Bhat, Prashant
3
; Antoshechkin, Igor A.
4
and Kitchen, Sheila A.
5
1✉ Division of Biology and Biological Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, United States of America.
2Department of Biology, DeSales University, 2755 Station Avenue, Center Valley, PA 18034, United States of America.
3Division of Biology and Biological Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, United States of America & David Geffen School of Medicine, University of California - Los Angeles, 10833 Le Conte Avenue, Los Angeles, CA 90095, United States of America.
4Division of Biology and Biological Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, United States of America.
5Division of Biology and Biological Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, United States of America.
2022 - Volume: 62 Issue: 2 pages: 532-573
https://doi.org/10.24349/pjye-gkeoOriginal research
Keywords
Abstract
Introduction
In the past couple of years, the number of sequenced animal genomes has increased dramatically, especially for arthropods about 500 genomes sequences are now available (Childers, 2020; Thomas et al., 2020). The majority of these genomes, however, belong to the insects (e.g. flies, beetles, wasp, butterflies and bugs (Thomas et al., 2020)) which compromise the most diverse, yet evolutionarily young and more derived taxa of arthropods (Regier et al., 2010; Giribet and Edgecombe, 2019). In strong contrast, genome assemblies, many of which are incomplete or not well annotated, exist for the Chelicerata (Childers, 2020) – the other major subphylum of arthropods (Regier et al., 2010; Giribet and Edgecombe, 2019). Chelicerates include sea spiders, spiders, mites and scorpions among other organisms, as well as several extinct taxa (Dunlop and Selden, 1998; Ballesteros and Sharma, 2019). Chelicerates originated as marine animals about 500 million years ago (Dunlop and Selden, 1998; Dunlop, 2010). Molecular analyses suggest that one particular group, the omnivorous and detritivores acariform mites, may have been among the first arthropods that colonized terrestrial habitats and gave rise to ancient, simple terrestrial food webs (Walter and Proctor, 1999; Dunlop and Alberti, 2008; Schaefer et al., 2010).
So far, the well-annotated genomic data of chelicerates is limited to animal parasites (including human pathogens and ticks), plant parasites, and predatory mites used in pest control (Cornman et al., 2010; Grbić et al., 2011; Rider et al., 2015; Gulia-Nuss et al., 2016; Hoy et al., 2016; Dong et al., 2017; Dong et al., 2018). Other than some lower-quality genome assemblies (Bast et al., 2016), there are no resources available for free-living soil and litter inhabiting species. Such data are, however, pivotal to understanding the evolution of parasitic lifestyles from a free-living condition and to bridge the gap between early aquatic chelicerates such as horseshoe crabs, and highly derived terrestrial pest species and parasites (Klimov and OConnor, 2013; Weinstein and Kuris, 2016; Shingate et al., 2020). Because the phylogeny of Chelicerata remains unresolved, additional chelicerate genomes are urgently needed for comparative analyses (Dunlop, 2010; Ballesteros and Sharma, 2019; Lozano-Fernandez et al., 2019). To help address this deficit, we report here the genome assembly of the soil dwelling oribatid mite Archegozetes longisetosus (Aoki, 1965; Figure 1) (Aoki, 1965) and a comprehensive analysis in the context of developmental genes, feeding biology, horizontal gene transfer and biochemical pathway evolution of chelicerates.

Archegozetes longisetosus (hereafter referred to as Archegozetes) is a member of the Oribatida (Acariformes, Sarcoptiformes), an order of chelicerates well-known for their exceptional biosynthesis capacities, biochemical diversity, unusual mode of reproduction, unusually high pulling strength, mechanical resistances and pivotal ecological importance (Norton and Palmer, 1991; Maraun and Scheu, 2000; Heethoff and Koerner, 2007; Maraun et al., 2007; Heethoff et al., 2009; Raspotnig, 2009; Brückner et al., 2017b; Schmelzle and Blüthgen, 2019; Brückner et al., 2020). Archegozetes, like all members of its family Trhypochthoniidae (Figure 1a), reproduce via thelytoky (Heethoff et al., 2013). That means the all-female lineages procreate via automictic parthenogenesis with an inverted meiosis of the holokinetic chromosomes, resulting in clonal offspring (Palmer and Norton, 1992; Wrensch et al., 1994; Heethoff et al., 2006; Bergmann et al., 2018). While studying a parthenogenetic species is useful for the development of genetic tools as stable germ-line modifications can be obtained from the clonal progeny without laboratory crosses, one is confronted with the technical and philosophical problems of species delineation, cryptic diversity and uncertain species distribution (Heethoff et al., 2013; Oxley et al., 2014). Reviewing all available data, Norton (Norton, 1994; 2007) and Heethoff et al. (2013) concluded that Archegozetes is found widely on continents and islands throughout the tropical and partly subtropical regions of the world and that it is a middle-derived oribatid mite closely related to the suborder Astigmata.
One major feature of most oribatid mites is a pair of opisthonotal oil-glands and Archegozetes is no exception (Sakata and Norton, 2001; Raspotnig, 2009). These are a pair of large exocrine glands, each composed of a single-cell layer invagination of the cuticle, which is the simplest possible paradigm of an animal gland (Heethoff, 2012; Brückner and Parker, 2020). The biological role of these glands was rather speculative for a long time; ideas ranged from a lubricating and osmo- or thermoregulative function (Zachvatkin, 1941; Riha, 1951; Smrž, 1992) to roles in chemical communication (Shimano et al., 2002; Raspotnig, 2006; Heethoff et al., 2011a). So far about 150 different gland components have been identified from oribatid mites, including mono- and sesquiterpenes, aldehydes, esters, aromatics, short-chained hydrocarbons, hydrogen cyanide (HCN) and alkaloids (Saporito et al., 2007; Raspotnig, 2009; Brückner et al., 2015; Brückner et al., 2017b; Heethoff et al., 2018). While some chemicals appear to be alarm pheromones (Shimano et al., 2002; Raspotnig, 2006), most function as defensive allomones (Heethoff et al., 2011a). Interestingly, alkaloids produced by oribatids mites are the ultimate source of most toxins sequestered by poison-frogs (Saporito et al., 2007; Saporito et al., 2009).
Terrestrial chelicerates predominately ingest fluid food. While phloem-feeding plant pests like spider mites and ecotoparasites like ticks adapted a sucking feeding mode, scorpions, spiders and others use external, pre-oral digestion before ingestion by morphologically diverse mouthparts (Cohen, 1995; Dunlop and Alberti, 2008; Bensoussan et al., 2016; Gulia-Nuss et al., 2016). Exceptions from this are Opiliones and sarcoptiform mites i.e., oribatid and astigmatid mites, all of which ingest solid food (Norton, 2007; Shultz, 2007; Heethoff and Norton, 2009). In general, oribatids feed on a wide range of different resources and show a low degree of dietary specialization (Brückner et al., 2018b). The typical food spectrum of Oribatida, includes leaf-litter, algae, fungi, lichens, nematodes, and small dead arthropods such as collembolans (Riha, 1951; Schneider et al., 2004a; Schneider et al., 2004b; Schneider and Maraun, 2005; Heidemann et al., 2011). In laboratory feeding trials, oribatid mites tend to prefer dark pigmented fungi, but also fatty acid-rich plant-based food (Schneider and Maraun, 2005; Brückner et al., 2018b). Additionally, stable-isotope analyses of 15N and 13C suggested that Oribatida are primary- and secondary decomposers feeding on dead plant material and fungi, respectively (Schneider et al., 2004a; Maraun et al., 2011). The reasons for these preferences are still unknown, but they raise the question of how oribatid mites are able to enzymatically digest the cell walls of plants and fungi (Schneider et al., 2004b; Smrž and Čatská, 2010; Brückner et al., 2018a; Brückner et al., 2018b).
Early studies on Archegozetes and other mites found evidence for cellulase, chitinase and trehalase activity which was later attributed to symbiotic gut bacteria (Zinkler, 1971; Luxton, 1979; Haq, 1993; Siepel and de Ruiter-Dijkman, 1993; Smrž, 2000; Smrž and Norton, 2004; Smrž and Čatská, 2010). While such bacterial symbionts are a possible explanation, genomic data of other soil organisms and plant-feeding arthropods suggest a high frequency of horizontal transfer of bacterial and fungal genes enabling the digestion of cell walls (Grbić et al., 2011; Mayer et al., 2011; Wybouw et al., 2016; Wu et al., 2017; Wybouw et al., 2018; McKenna et al., 2019). For instance, an in-depth analysis of the spider mite Tetranychus urticae revealed a massive incorporation of microbial genes into the mite's genome (Grbić et al., 2011; Wybouw et al., 2018). Horizontal gene transfer appears to be a common mechanism for soil organisms, including mites, to acquire novel metabolic enzymes (Hoffmann et al., 1998; Grbić et al., 2011; Mayer et al., 2011; Faddeeva-Vakhrusheva et al., 2016; Wu et al., 2017; Dong et al., 2018), and hence seems very likely for Archegozetes and other oribatid mite species that feed on plant or fungal matter.
Archegozetes has been established as a laboratory model organism for three decades, having been used in studies, ranging from ecology, morphology, development and eco-toxicology to physiology and biochemistry (Barnett and Thomas, 2012; 2013b; 2013a; Heethoff et al., 2013; Brückner et al., 2017a; Barnett and Thomas, 2018; Brückner et al., 2020). As such, Archegozetes is among the few experimentally tractable soil organisms and by far the best-studied oribatid mite species (Thomas, 2002; Barnett and Thomas, 2012; Heethoff et al., 2013). Since it meets the most desirable requirements for model organisms (Thomas, 2002), that is a rapid development under laboratory conditions, a dedicated laboratory strain was named Archegozetes longisetosus ran in reference to its founder Roy A. Norton (Heethoff et al., 2013, Figure 1b-c). Their large number of offspring enables mass cultures of hundreds of thousands of individuals, and their cuticular transparency during juvenile stages, and weak sclerotization as adults are general assets of an amenable model system (Heethoff and Raspotnig, 2012; Heethoff et al., 2013; Brückner et al., 2016; Brückner et al., 2018c). In the past 10 years, Archegozetes also received attention as a model system for chemical ecology (Raspotnig et al., 2011; Heethoff and Raspotnig, 2012; Heethoff and Rall, 2015; Brückner et al., 2016; Brückner and Heethoff, 2018; Thiel et al., 2018; Brückner et al., 2020). Some of these studies focusing on the Archegozetes gland revealed basic insights into the chemical ecology and biochemical capabilities of arthropods (Heethoff and Rall, 2015; Thiel et al., 2018; Brückner et al., 2020). Hence, Archegozetes is poised to become a genetically tractable model to study the molecular basis of gland and metabolic biology.
The aim and focus of the current study were three-fold – to provide well-annotated, high-quality genomic and transcriptomic resources for Archegozetes longisetosus (Figure 1), to reveal possible horizontal gene transfers that could further explain the feeding biology of oribatids, and to present Archegozetes as a research model for biochemical pathway evolution. Through a combination of comparative genomic and detailed computational analyses, we were able to generate a comprehensive genome of Archegozetes and provide it as an open resource for genomic, developmental and evolutionary research. We further identified candidate horizontal gene transfer events from bacteria and fungi that are mainly related to carbohydrate metabolism and cellulose digestion, features correlated with the mite feeding biology. We also used the genomic data together with stable-isotope labeling experiments and mass spectrometric investigation to delineate the biosynthesis pathway of monoterpenes in oribatid mites.
Results and discussion
Archegozetes longisetosus genome assembly
Archegozetes longisetosus (Figure 1) has a diploid chromosome number (2n) of 18 (Heethoff et al., 2006), most likely comprising 9 autosomal pairs, the typical number of nearly all studied oribatid mite species (Figure 1d) (Norton et al., 1993). There are no distinct sex chromosomes in Archegozetes; this appears to be ancestral in the Acariformes and persisted in the Oribatida (Norton et al., 1993; Wrensch et al., 1994; Heethoff et al., 2006). Even though some XX:XO and XX:XY genetic systems have been described in the closely related Astigmata, the sex determination mechanism in oribatids, including Archegozetes, remains unknown (Oliver Jr, 1983; Norton et al., 1993; Wrensch et al., 1994; Heethoff et al., 2006; Heethoff et al., 2013).
To provide genetic resources, we sequenced and assembled the genome using both Illumina short-read and Nanopore MinION long-read sequencing approaches followed by scaffolding with Hi-C technology (Figure 1d; Table 1; see also ''Materials and Methods''). Analyses of the k-mer frequency distribution of short reads (Table 1; Supplementary Figure S1) resulted in an estimated genome size range of 135-180 Mb, encompassing the final HiC assembled size of 143 Mb (Table 1; see also ''Materials and Methods''). Compared to genome assemblies of other acariform mites, the assembled genome size of Archegozetes is on the large end of the spectrum (with Opiella nova and O. subpectinata representing notable exceptions)(Brandt et al., 2021), but is smaller than that of most mesostigmatid mites, ticks and spiders that on average range from ~250 Mb up to 2.5 Gb (Grbić et al., 2011; Bast et al., 2016; Gulia-Nuss et al., 2016; Hoy et al., 2016; Dong et al., 2017; Schwager et al., 2017; Dong et al., 2018). In the context of arthropods in general, Archegozetes' genome (Table 1) is among the smaller ones and shares this feature with other arthropod model species like Tetranychus urticae (hereafter called'spider mite'), Drosophila, clonal raider ant and red flour beetle (Consortium, 2008; Grbić et al., 2011; Oxley et al., 2014; dos Santos et al., 2015). Even though we surface-washed the mites and only used specimens with empty alimentary tracts for sequencing, we removed a total 532 sequences (438 long-read contigs and 94 Hi-C scaffolds) with high bacterial or fugal homology making up approximately 9 Mb of contamination in the final assembly (see supplementary Table S1). The final filtered Hi-C genome assembly was composed of 164 scaffolds, the majority of which is composed of nine pseudochromosomes with an N50 contiguity of 16.25 Mb (Table 1 and Figure 1d).


Genome scaffolding and the analysis of genome structure in arthropods by all-vs-all chromosome conformation capture (Hi-C) is a relatively new field that has grown in recent years (Richards, 2019). Like other arthropods, the Archegozetes genome is organized into chromosome territories, compartments, and sub-compartment structures (Figure 1d). Classic microscopy and fluorescent staining previously revealed that Archegozetes has a diploid set of 18 (2n) highly condensed chromosomes, and we recovered nine pseudo-chromosomes in our haploid assembly (Heethoff et al., 2006). Even though Hi-C is often not able to resolve inter-chromosomal interactions and long range-contacts (Quinodoz et al., 2022), we discovered multiple such contacts between different chromosomes, for instance chromosome 1 is interacting with parts of each chromosome except chromosome 5 (Figure 1d). One hypothesis which could explain this pattern is that the Archegozetes genome is highly compact, and chromatin is densely packed in the nucleus (see also the genome of the tomato russet mite; Greenhalgh et al., 2020).
The official gene set and annotation of Archegozetes

We generated the official gene set (OGS) for Archegozetes by an automated, multi-stage process combining ab inito and evidenced-based (RNAseq reads, transcriptomic data and curated protein sequences) gene prediction approaches (see ''Materials and Methods'') yielding 24,538 gene models. In comparison to other mites and ticks as well as insects, this is well within the range of the numbers discovered in other Chelicerata so far (Figure 2a). Chelicerates with a large OGS, however, usually possess larger genomes (1-7 Gb), which suggests that Archegozetes may have a relatively dense distribution of protein-coding genes in its genome. On the other hand, ticks can have giga-base sized genomes, but only a rather small number of gene models, probably due to high repetitive content (Palmer et al., 1994; Van Zee et al., 2007; Gulia-Nuss et al., 2016; Barrero et al., 2017). Lacking more high-quality genomic resources of mites, it is thus not clear whether the OGS of Archegozetes is the rule, or rather the exception within the Oribatida.
To compare if Archegozetes' OGS is similar to predicted genes of other oribatid mites as well as Prostigmata and Astigmata, we first clustered genes by ortholog inference (OrthoFinder; Emms and Kelly (2015)), removed species-specific genes and constructed a presence-absence matrix of orthogroups to ordinate the data using non-metric multidimension scaling (NMDS, Figure 2b). Ordination revealed that the OGS of Archegozetes is well nested with other oribatid mites and clearly separated from their closest relative the astigmatid mites as well as prostigmatid mites (Figure 2b). As a first step in annotating the OGS, we ran KOALA (KEGG Orthology And Links Annotation) to functionally characterize the genes (Kanehisa et al., 2016). In total, 9,719 (39.6%) of all genes received annotation and about two thirds of all genes were assigned either as genes related to genetic information processing (34%) or metabolic genes (30%), while the remaining genes fell into different KEGG categories (Figure 2c). To further annotate the genome, we followed the general workflow of funannotate with some modifications (Palmer and Stajich, 2017, see ''Materials and Methods'').
Overall, we found 16,685 genes (68%) of the OGS with homology to previously published sequences (Figure 2d). For over half of all genes (56%), we were able to assign a full annotation, 4% of all genes only showed homology to bioinformatically predicted proteins of other species, while 8% of all genes only showed homology to hypothetical proteins (Figure 2d). As only a few high-quality, annotated mite genomes are available and the two-spotted spider mite is the sole species with any experimentally confirmed gene models, it is not surprising that we were only able to confidently annotate about 60% of all genes of the OGS (Figure 2d).
Orthology and comparative genomics of chelicerates

To further access the protein-coding genes of the mite, we compared the OGS to other chelicerates. Both concatenated maximum likelihood and coalescent species-tree phylogenomic approaches based on 1,121 orthologs placed Archegozetes, as expected, within the Nothrina (Pachl et al., 2012; Heethoff et al., 2013) with strong support and recovered previously found oribatid clade topologies (Figure 3a). Our analysis placed the Astigmata as a sister group of Oribatida and not nested within oribatids as suggested based on life-history, chemical defensive secretions, morphology and several molecular studies (Norton, 1994; 1998; Sakata and Norton, 2001; Alberti and Michalik, 2004; Maraun et al., 2004; Liana and Witaliński, 2005; Domes et al., 2007; Dabert et al., 2010; Koller et al., 2012; Pepato and Klimov, 2015; Klimov et al., 2018; Li and Xue, 2019). The relationship of Oribatida and Astigmata has been challenging to resolve for the past decades and several studies using different set of genes, ultra-conserved elements or transcriptomic data reconstructed discordant phylogenies, some of which are similar to ours (Maraun et al., 2004; Domes et al., 2007; Dabert et al., 2010; Pepato and Klimov, 2015; Klimov et al., 2018; Li and Xue, 2019; Lozano-Fernandez et al., 2019; Van Dam et al., 2019). Overall, the oribatid-astigmatid relationship remains unresolved and a broader taxon sampling, especially of more basal Astigmata, will be necessary (Norton, 1994; 1998; Domes et al., 2007; Klimov et al., 2018; Lozano-Fernandez et al., 2019; Van Dam et al., 2019). We recovered Trombidiformes (Prostigmata and Sphaerolichida) as sister group of the Sarcoptiformes (Oribatida and Astigmata) constituting the Acariformes (Figure 3a). Neither the maximum likelihood phylogeny (Figure 3a), nor the coalescence-based phylogeny (Supplementary Figure S2) reconstructed the Acari (i.e., Acariformes and Parasitiformes) as a monophyletic clade. Even though there is morphological, ultrastructural and molecular evidence for a biphyletic Acari, as we recovered here, this relationship and larger-scale chelicerate relationships remain unclear (Alberti, 1984; 1991; Dabert, 2006; Dunlop and Alberti, 2008; Jeyaprakash and Hoy, 2009; Li and Xue, 2019; Lozano-Fernandez et al., 2019; Van Dam et al., 2019).
To further assess the quality and homology of both the genome assembly (Table 1) and the OGS (Figure 2), we used the 1066 arthropod Benchmarking Universal Single-Copy Ortholog (BUSCO) genes data set (Simão et al., 2015). Nearly all BUSCO genes were present in the Archegozetes assembly and OGS (94.6% and 95.4%, respectively; Figure 3b). Compared to other genomes sequenced so far, the Archegozetes genome has the highest completeness among oribatid mites and the OGS completeness is on par to the well curated genomes of other chelicerate species and Drosophila melanogaster (Figure 3b). This result is not surprising because the Archegozetes genome was assembled from long-read and short-read data followed by scaffolding with Hi-C technology, while most other oribatid mite genomes were solely short reads sequenced on older Illumina platforms (Bast et al., 2016). The fraction of duplicated BUSCO genes in Archegozetes (1.2%) was smaller than the spider mite and deer tick (Grbić et al., 2011; Gulia-Nuss et al., 2016), but very low compared to the house spider (Figure 3c), whose genome underwent an ancient whole-genome duplication (Schwager et al., 2017).
Overall, the high quality of both the genome assembly and OGS of Archegozetes is unprecedented as compared to genomic resources of other oribatid mites. We next categorized all protein models from the OGS by conversation level based on a global clustering orthology analysis (OrthoFinder; Emms and Kelly, 2015) of 23 species (Figure 3c; supplementary Figure S3) representing Acariformes, Parasitiformes, several other chelicerates and the fly Drosophila. As for most other species (Siepel et al., 2005; Thomas et al., 2020), about a third of all orthogroups was highly conserved (Figure 3c) across the arthropods, being either in all species (10%; Figure 3d) or is most (22%; Figure 3d). Only 1% of all Archegozetes orthogroups did not show homology and were species specific (Figure 3c and d). Only a low proportion (Figure 3c) of orthogroups was conserved across the higher taxonomic levels (all \textless1% in Archegozetes; Figure 3d), which is in line with previous studies that included prostigmatid and mesostigmatid mites (Hoy et al., 2016; Dong et al., 2017; Dong et al., 2018). Interestingly, there was a large proportion of orthogroups conserved across all Oribatida (43% in Archegozetes; Figure 3d) and also about 19% of orthogroups in Archegozetes were shared only with other Nothrina (Figure 3d). A fairly large percentage of these orthogroups may contain potentially novel genes that await experimental verification and functional analyses (Emms and Kelly, 2015; Nagy et al., 2020; Thomas et al., 2020). Especially the lack of homology within the Sarcoptiformes (2-3%; Figure 3c) may explain the controversial placement of Astigmata as a sistergroup of Oribatida that we recovered (Figure 3a). This grouping is likely caused by a long-branch attraction artifact and the sister relationship was incorrectly inferred (Dabert, 2006; Domes et al., 2007; Dabert et al., 2010; Pepato and Klimov, 2015; Klimov et al., 2018), because orthogroup clustering could not detect enough homology between oribatids and the Astigmata so far sequenced, which are highly derived. Hence, a broad taxon sampling of basal astigmatid mite genomes seems necessary to resolve Oribatida-Astigmata relationship (Norton, 1994; 1998; Pepato and Klimov, 2015; Li and Xue, 2019; Van Dam et al., 2019).
Repeat content analysis and transposable elements (TEs)
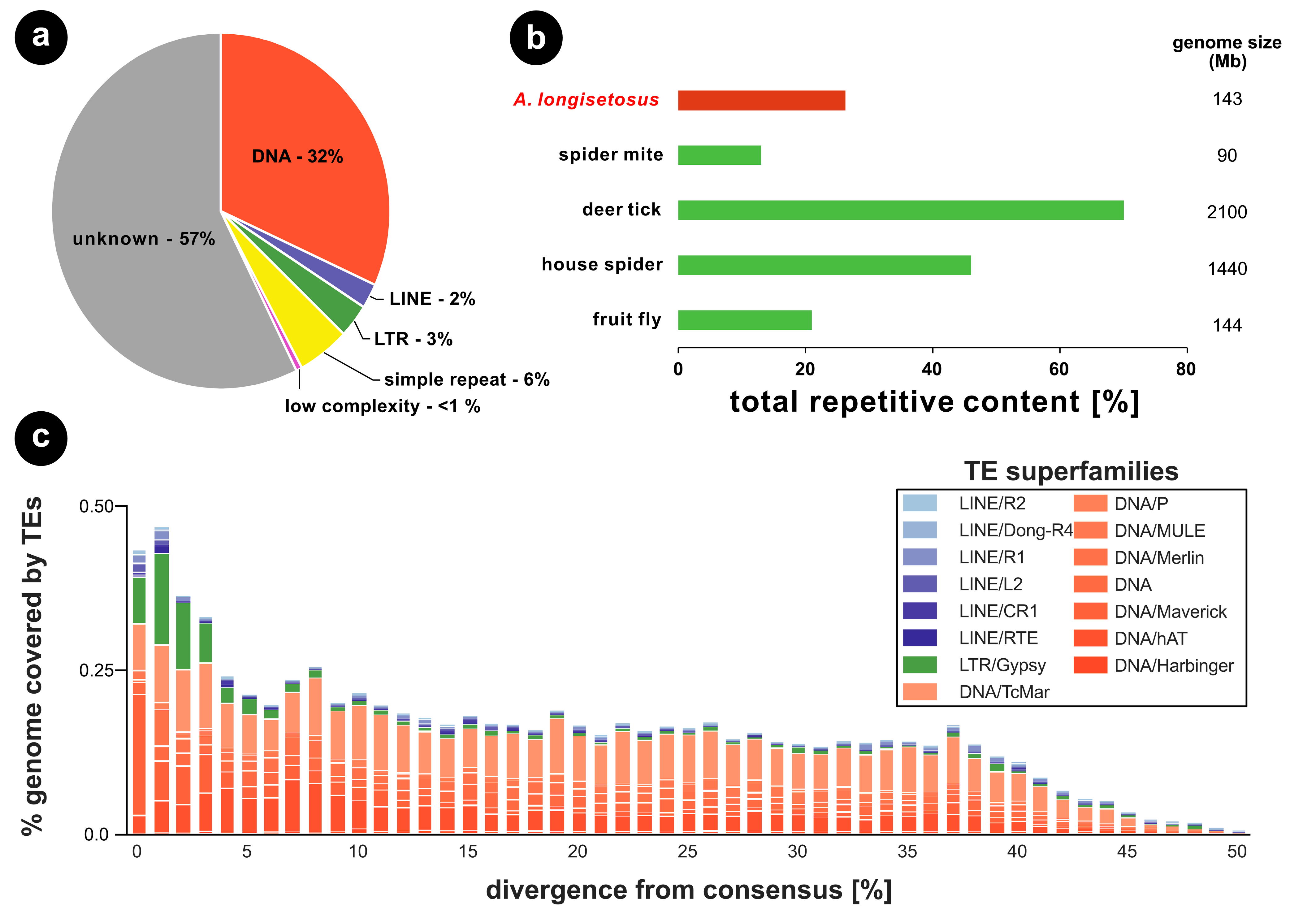
For clonal species like Archegozetes, reproducing in the absence of recombination, it has been hypothesized that a reduced efficacy of selection could results in an accumulation of deleterious mutations and repeats in the genome (Muller, 1964; Arkhipova and Meselson, 2000; Nuzhdin and Petrov, 2003; Schön et al., 2009; Barton, 2010; Charlesworth, 2012). There is, however, no evidence for such an accumulation in oribatids or other arthropods (Bast et al., 2016). The genome of Archegozetes repeat content was predicted to be 26% of the genome (Figure 4) Generally, we found that most of the repetitive content in Archegozetes could not be classified (57%; Figure 4a). The high proportion of unknown repeats likely corresponds to novel predicted repetitive content, because of limited repeat annotation of mites in common repeat databases such as RepBase (Bast et al., 2016). Regarding the two major classes of repeat content, DNA transposons made up about 32% of total repeats, while only 5% represented retrotransposons (Figure 4a). About 6% of total repetitive content comprised simple and low complexity repeats (Figure 4a). Overall, the total repetitive content (26%, Figure 4b) seems to be within a normal range for chelicerates and arthropods.
The repeat content found in other oribatid mites was lower (Bast et al., 2016), but recent studies suggest that sequencing technology, read depth and assembly quality are paramount to the capacity of identifying repeat content and TEs (Bourque et al., 2018; Panfilio et al., 2019). Hence, it is very likely the current genomic data for other Oribatida underestimate the actual total repetitive content. More low-coverage, long-read sequencing could reduce the assembly fragmentation and likely reveal a higher proportion of repeats, closer to the actual repetitiveness of oribatid genomes (Panfilio et al., 2019).
Different classes of transposable elements (TEs) are characterized by the mechanism they use to spread within genomes and are known to influence population dynamics differently (Finnegan, 1989; Bourque et al., 2018; Crescente et al., 2018). We therefore analyzed the evolutionary history of TE activity in Archegozetes in more detail (Figure 4c). The main TE superfamilies were DNA transposons (Figure 4a and c), which seems to be a common pattern of oribatid mite genomes. For Archegozetes, they appear to have accumulated in the genome for a long time (i.e. they are more divergent from the consensus; (Waterston et al., 2002)) with Tc1/mariner – a superfamily of interspersed repeats DNA transposons (Bourque et al., 2018) – being the most abundant one (Figure 4c). Interestingly, we found an increase in TE activity with 0-4% sequence divergence range, indicating a recent burst (Figure 4c). This burst contained an enrichment of DNA Mavericks, which are the largest and most complex DNA transposons with homology to viral proteins (Bourque et al., 2018), but also several of retrotransposons. Among these, is the Long Terminal Repeat (LTR) gypsy retroelement (Figure 4c), which is closely related to retroviruses (Bourque et al., 2018). Like retroviruses, it encodes genes equivalent to gag, pol and env, but relatively little is known about how it inserts its DNA into the host genome (Dej et al., 1998; Havecker et al., 2004). So far, it is unknown what these TEs do in Archegozetes, but the recent burst in TE abundance might suggest that some changes in the genome might have happened since the became a laboratory model nearly 30 years ago (Heethoff et al., 2013).
The Archegozetes Hox cluster
The Hox genes are a group of highly conserved transcription factor-encoding genes that are used to pattern the antero-posterior axis in bilaterian metazoans (Holland and Hogan, 1988; Hrycaj and Wellik, 2016). Ancestrally, arthropods likely had ten Hox genes arranged in a cluster (Hughes and Kaufman, 2002). During arthropod development, the Hox genes specify the identities of the body segments, and mutations in Hox genes usually result in the transformation of segmental identities (Hughes and Kaufman, 2002). The importance of Hox genes in development of metazoans makes knowledge of their duplication and disappearances important for understanding their role in the evolution of body plans (Hughes and Kaufman, 2002).
Mites largely lack overt, external signs of segmentation, other than the serially arranged appendages of the prosoma (Dunlop and Lamsdell, 2017). Signs of segmentation in the posterior body tagma, the opisthosoma, do exist in adult members of Endeostigmata (van der Hammen, 1970). However, these segmental boundaries are largely present only in the dorsal opisthosoma, making it difficult to assess how these correspond to the ventral somites (van der Hammen, 1970; Dunlop and Lamsdell, 2017). Developmental genetic studies of the spider mite and Archegozetes suggest that acariform mites only pattern two segments in the posterior body region, during embryogenesis (Grbić et al., 2011; Barnett and Thomas, 2012; 2013b; 2018). This stands in stark contrast to other studied chelicerate embryos. For example, during embryogenesis the spider Parasteatoda tepidariorum patterns twelve opisthosomal segments (Schwager et al., 2015) and the opilionid Phalangium opilio patterns seven (Sharma et al., 2012). Furthermore, a member of Parasitiformes, the tick Rhipicephalus microplus, appears to pattern eight opisthosomal segments during embryogenesis (Santos et al., 2013).
Parallel to the observation of segmental reduction in the spider mite, genomic evidence suggests that this acariform mite has lost two of its Hox genes, i.e., Hox3 and abdominal-A (abd-A) (Grbić et al., 2011). Interestingly, orthologs of abd-A in other studied arthropods pattern the posterior segments as well. A genomic comparison of arthropod Hox clusters has also shown a correlation between independent losses of abd-A and a reduction in posterior segmentation (Pace et al., 2016). To investigate whether the loss of segmentation in Archegozetes is also due to an absence in abd-A, we annotated its Hox cluster, paying close attention to the region between the Hox genes Ultrabithorax (Ubx) and Abdominal-B (Abd-B), which is usually where this gene resides in other arthropods (Hughes and Kaufman, 2002). Our results suggest that the Archegozetes Hox genes are clustered in a contiguous sequence (HiC scaffold 3, total size ~12.36 Mbp) in the same order as suggested for the ancestral arthropod (Heethoff and Rall, 2015). Furthermore, we found no sequences suggestive of an abd-A ortholog in Archegozetes (Figure 5a). These data also support the findings of a previous PCR survey that retrieved no abd-A ortholog in Archegozetes (Cook et al., 2001). Genomic evidence from the Parasitiformes Ixodes scapularis and Metaseiulus occidentalis reveal that these taxa maintain orthologs of all ten Hox genes, however in M. occidentalis these genes are not clustered as they are in I. scapularis (Gulia-Nuss et al., 2016; Hoy et al., 2016).
Taken together, these observations suggest that the last common ancestor of acariform mites likely lost its abdominal-A gene as well as experiencied a reduction in opisthosomal segmentation (Figure 5b). Alternatively, these shared losses of abd-A may be due to convergence due to similar selective pressures favoring a reduction in body size. The dorsal, external segmentation of endeostigmatid mites does not necessarily contradict the hypothesis of a loss of abd-A at the base of the acariform mites. As Hox genes are usually deployed after the genetic establishment of segments in arthropods (Hughes and Kaufman, 2002), the opisthosomal segments in endeostigmatid mites may still develop in the absence of abd-A. However, this hypothesis needs further testing with observations of segmental gene expression in endeostigmatids as well as additional acariform species.

Life-stage specific RNA expression patterns
Developmental and gene expression data from Archegozetes embryos (Figure 5 d and e) have elucidated many of the potential mechanisms driving the morphogenesis of many developmental peculiarities. These peculiarities include the suppression of the fourth pair of walking legs during embryogenesis as well as the reduction of opisthosomal segmentation (Telford and Thomas, 1998; Thomas, 2002; Barnett and Thomas, 2012; 2013a; 2013b; 2018). In typical acariform mites, embryogenesis ends with the first instar, the prelarva, which usually remains within the egg chorion, as in Archegozetes. Hatching releases the second instar, the larva, which is followed by three nymphal instars (proto-, deutero- and tritonymph) and the adult, for a total of six instars. (Heethoff et al., 2007). Thus far, methodological limitations have made it difficult to examine how mite segmentation and limb development progress throughout these instars.
To this end, we used RNAseq to calculate the transcripts per million (tpm) values of genes known to be, or suspected to be, involved in limb development and segmentation throughout the six different instars of Archegozetes. Prior to comparing these tpm values, gene orthology was confirmed via phylogenetic analyses (supplementary Figures S4-S11; see Table S2 for phylogenetic statistics and Table S3 for tpm values). Regarding the total number of genes expressed across the different life stages, we found that earlier instars generally expressed a higher number of genes (Figure 5c). While most expressed genes were shared across all instars, more transcripts were shared between the eggs and the larvae and among all five juvenile instars. Additionally, we found that earlier instars expressed a larger number of stage-specific genes as compared to later instars and adults (Figure 5c).
Gene expression, scanning electron microscopy (SEM) and time-lapse data have revealed that the development of the fourth pair of walking legs in Archegozetes is suppressed until after the larval instar (Telford and Thomas, 1998; Barnett and Thomas, 2012; 2018). The resulting larva is thus hexapodal (see also embryo in Figure 5e), which constitutes a putative synapomorphy of Acari, if they are monophyletic (Dunlop and Alberti, 2008). In arthropods, the development of the limbs is generally accomplished via the activity of highly conserved regulatory genes, termed the ''limb gap genes.'' These genes are expressed along their proximo-distal axes to establish the specific identities of the limb podomeres. The limb gap genes include extradenticle (exd) and homothorax (hth), which act together to specify the proximal limb podomeres, dachshund (dac), which specifies the medial podomeres, and Distal-less (Dll) which specifies the distal-most podomeres. It was previously shown that the deployment of these genes in the anterior appendages of Archegozetes, i.e., the chelicerae, pedipalps and first three pairs of walking legs (Figure 5d and e), is similar to that of other chelicerate taxa (Barnett and Thomas, 2013a; Schwager et al., 2015; Sharma et al., 2015). However, in the anlagen of the fourth pair of walking legs, only the proximal-specifying genes, exd and hth, are expressed (Barnett and Thomas, 2013a).
Whether the limb gap genes are re-deployed during the transition from the prelarval to larval instars in order to activate the development of the fourth pair of walking legs remains an open question. We therefore compared the average tpm values of verified limb gap genes (i.e., Al-Dll, Al-Hth, Al-exd, and Al-dac (Barnett and Thomas, 2013a)) in embryos and at each instar stage (Figure 5f). We also compared the tpm values of the Archegozetes orthologs of Sp6-9 and optomotor blind, genes shown to be involved in limb formation in spiders (Königsmann et al., 2017; Heingård et al., 2019). We hypothesized that limb development genes would show high expression in the larval stage leading to the development of the octopodal protonymph. We did observe an increase in the tpm averages of Al-hth as well as Al-optomotor-blind, however the aforementioned limb gap gene expression levels were similar between these instars (Figure 5f). Taken together, these genes may not be up-regulated for the formation of the fourth pair of walking legs between these two instars.
Chelicerate embryos segment their bodies through a ''short/intermediate germ'' mechanism, whereby the anterior (prosomal) segments are specified asynchronously (Schwager et al., 2015). This usually occurs well before the sequential addition of posterior segments from a posterior growth zone. Based on neontological and paleontological data, chelicerate arthropods may have ancestrally had an opisthosoma comprised of 12 or more segments (Dunlop and Selden, 1998; Dunlop, 2010; Dunlop and Lamsdell, 2017). Embryonic expression data for the segment polarity genes, those genes that delineate the boundaries of the final body segments, have shown that in most studied chelicerate embryos opisthosomal segments are delineated during embryogenesis (Schwager et al., 2015; Dunlop and Lamsdell, 2017). However, as discussed above, expression data in Archegozetes embryos suggest that only two opisthosomal segments are patterned during embryogenesis (Barnett and Thomas, 2012; 2018); this indicates that mites have significantly reduced their number of opisthosomal segments either by loss or by fusion. Further complicating this is the observation that many mites add segments as they progress through the larval instars, a phenomenon known as anamorphic growth (Dunlop and Lamsdell, 2017).
To determine by what genetic process Archegozetes may add segments during post-embryonic ontogeny, we assessed the expression of known chelicerate and arthropod segmentation genes in each instar transcriptome (Figure 5f) (Schwager et al., 2015). We observed an up-regulation of the segmentation genes hedgehog and engrailed in the larvae, as well as the slight up-regulation of patched and pax3/7. Furthermore, the segmentation gene wingless was slightly up-regulated in the protonymph, as well as a slight up-regulation of hedgehog in the tritonymph. Lastly, we found that transcripts of the genes pax3/7 and runt were up-regulated in adults. These results suggest that Archegozetes does pattern body segments during the progression through the it's instars similar to other Chelicerata (Dunlop and Lamsdell, 2017).
Another peculiarity of Archegozetes is that these mites lack eyes (see more details below). Eye loss has been documented in other arachnid clades, including independently in other members of Acari (Evans, 1992; Walter and Proctor, 1999), and it has been recently demonstrated that a species of whip spider has reduced its eyes by reducing the expression of retinal determination genes that are shared throughout arthropods (Gainett et al., 2020). We sought to determine if eye loss in Archegozetes also is associated with the reduced expression of these genes (see also analysis of photoreceptor genes below). The genes, which have been shown to be expressed in the developing eyes of spiders and whip scorpions, include Pax-6, six1/sine oculis (so), eyes absent (eya), Eyegone, Six3/Optix, and atonal (Samadi et al., 2015; Schomburg et al., 2015; Gainett et al., 2020). We also followed the expression of Al-orthodenticle, a gene previously shown to be expressed in the ocular segment of Archegozetes (Telford and Thomas, 1998). Surprisingly, all of these genes, excluding the Pax-6 isoform A and eyegone, are indeed expressed during embryogenesis (Figure 5f). Aside from the larval expression of the Pax-6 isoform A during the larval stage, these eye-development genes remain quiescent until the adult stage, where all but Pax-6 isoform A, six3 and atonal are up-regulated (Figure 5f). These results are exceedingly surprising, given the conserved role of genes in retinal patterning. They suggest a novel role for these genes, or alternatively, these expression patterns could be the result of early expression of a retinal determination pathway followed by negative regulation by other genes to suppress eye development.
Photoreceptor and chemosensory system of Archegozetes longisetosus

Unlike insects and crustaceans, chelicerates do not have compounds eyes – with horseshoe carbs being an exception. Generally, mites are eyeless or possess one or two pairs of simple ocelli (Patten, 1887; Exner, 1989; Alberti and Coons, 1999; Harzsch et al., 2006; Alberti and Moreno-Twose, 2012). Ocelli are common in Prostigmata and Endeostigmata, among Acariformes, as well Opilioacarida – the most likely sister group to the Parasitiformes – but are absent in most Oribatida, Astigmata, Mesostigmata and ticks (Walter and Proctor, 1998; Walter and Proctor, 1999; Norton and Fuangarworn, 2015; Norton and Franklin, 2018). This suggests that the presence of eyes might be an ancestral condition for both Acariformes and Parasitiformes, while more derived mites rely largely on chemical communication systems (Alberti and Coons, 1999).
In oribatid mites, detailed morphological and ultrastructural investigations have suggested that setiform sensilla are the most obvious sensory structures (Figure 6a) (Alberti, 1998; Alberti and Coons, 1999; Walter and Proctor, 1999). The trichobothria are very complex, highly modified (e.g., filiform, ciliate, pectinate, variously thickened or clubbed) no-pore setae which are anchored in a cup-like base and likely serve as mechanosensory structures. In contrast, the setal shafts of solenidia and eupathidia (Figure 6a) both possess pores (Alberti, 1998; Alberti and Coons, 1999; Walter and Proctor, 1999). Solenidia have transverse rows of small pores visible under a light microscope and likely function in olfaction, while the eupathidia have one or several terminal pores and likely are used as contact/gustatory sensilla (Figure 6a) (Alberti, 1998; Alberti and Coons, 1999). Previous work demonstrated that oribatid mites indeed use olfactory signals in the context of chemical communication and food selection (Shimano et al., 2002; Raspotnig, 2006; Heethoff et al., 2011a; Heethoff and Raspotnig, 2012; Brückner et al., 2018a; Brückner et al., 2018b).
Interestingly, detailed morphological and ultrastructural studies showed that light-sensitive organs exist in some Palaeosomata and Enarthronota (probably true eyes) as well as in Brachypylina (the secondary lenticulus), representing lower and highly derived oribatid mites, respectively (Alberti and Coons, 1999; Alberti and Moreno-Twose, 2012; Norton and Fuangarworn, 2015; Norton and Franklin, 2018). Archegozetes and most other oribatids, however, are eyeless, yet there is scattered experimental and some anecdotal evidence that even these mites show some response to light and seem to avoid it (`negative phototropism' or'negative phototaxis') (Trägårdh, 1933; Madge, 1965; Woodring, 1966; Walter and Proctor, 1999). Hence, we mined the genome of Archegozetes for potential photoreceptor genes and found one gene of the all-trans retinal peropsin class and one gene related to the spider mite rhodopsin-7-like gene (Figure 6b). Peropsin-like genes are also present in other eyeless ticks. In jumping spiders, they encode for nonvisual, photosensitive pigments, while rhodopsin-7 may be involved in basic insect circadian photoreception (Koyanagi et al., 2008; Nagata et al., 2010; Shen et al., 2011; Eriksson et al., 2013; Senthilan and Helfrich-Förster, 2016; Senthilan et al., 2019). Taken together, this might suggest that eyeless species like Archegozetes use peropsin- and rhodopsin-7-like genes for reproductive and diapause behaviors, or to maintain their circadian rhythm, as well as negative phototaxis.
The main sensory modality soil mites use is chemical communication via olfaction (Alberti, 1998; Alberti and Coons, 1999; Walter and Proctor, 1999; Raspotnig, 2006; Shen et al., 2011; Brückner et al., 2018a; Brückner et al., 2018b). In contrast to insects, but similar to crustaceans and Myriapoda, mites do not have the full repertoire of chemosensory classes, they are missing odorant receptors and odorant-binding proteins (Table 2) (Maraun et al., 2007; Raspotnig, 2009; Sánchez-Gracia et al., 2009; Sánchez-Gracia et al., 2011; Vieira and Rozas, 2011; Hoy et al., 2016; Dong et al., 2017; Dong et al., 2018). Although chemosensory protein (CSP) encoding genes are absent in most mite genomes, we identified one gene encoding for such a protein in Archegozetes and one CSP has been previously found in the deer tick (Table 2). Hence, Archegozetes should primarily rely on gustatory receptors (GRs) and ionotropic receptors (IRs). Both the number of GRs (44 genes; Figure 6d) and IRs (1 gene; Figure 6c) was very well within the range of most mites and ticks and there was no evidence for any massive chemoreceptor expansion like in the spider mite (Table 2) (Ngoc et al., 2016). This was surprising because Archegozetes, like other acariform mites have many multiporous solenidia, present on all legs and the palp, but appear to only have a limited number of chemoreceptors.

Canonical ionotropic glutamate receptors (iGluRs) are glutamate-gated ion channels with no direct role in chemosensation, which come in two major subtypes: either NMDA iGluRs which are sensitive to N-methyl-D-aspartic acid (NMDA) or non-NMDA iGluRs. The latter group – at least in Drosophila – seems to have essential functions in synaptic transmission in the nervous system and have been associated with sleep and vision (Benton et al., 2009; Sánchez-Gracia et al., 2009; Croset et al., 2010; Sánchez-Gracia et al., 2011; Ngoc et al., 2016). None of the IRs we found in the Archegozetes genome belonged to the NMDA iGluRs and all, but one were classified as non-NMDA iGluRs (Figure 6c). Nothing is known about their functions in mites. It is, however, likely that they perform similar tasks in synaptic transmission in the brain and musculature. In Drosophila a specific set of chemosensory IRs, which do not bind glutamate, respond to acids and amines (IR25a), but also to temperature (IR21a, IR93a). For Archegozetes we found one IR, similar to IR21a and IR93a of Drosophila, which fell into the antenna/1st leg IRs category (Table 2; Figure 6c) (Rytz et al., 2013; Knecht et al., 2016; Budelli et al., 2019). This is consistent with an assumed limited contribution of IRs to the perception of chemical cues. Furthermore, it is so far unclear whether this specific IR is expressed in the first pair of legs (Figure 6a and c) in Archegozetes, but similar genes seem to be expressed in the legs of other mite species (Dong et al., 2017; Dong et al., 2018), which could suggest a similar function as in the fruit fly.
GRs are multifunctional proteins and at least in insects they are responsible for the perception of taste, heat or volatile molecules (Montell, 2009). In Archegozetes we found 44 GRs, 20 of which had full RNAseq support across all life stages, yet none of them appeared to belong to a species-specific expansion of the GR gene family (Figure 6d). Generally, it is unclear if GRs in Archegozetes and other mites have similar functions as in insects, but the GR gene family is heavily expanded in many acariform mites and also is present in ticks (Table 2), suggesting an important biological role (Gulia-Nuss et al., 2016; Hoy et al., 2016; Ngoc et al., 2016; Barrero et al., 2017; Dong et al., 2017; Dong et al., 2018). This is supported by experimental evidence which suggested that ticks and other mites, including Archegozetes, use chemical cues to find their host, communicate or discriminate food (Yunker et al., 1992; Kuwahara, 2004; Raspotnig, 2006; Bunnell et al., 2011; Gulia-Nuss et al., 2016; Barrero et al., 2017; Brückner et al., 2018a; Brückner et al., 2018b).
In general, not much is known about the nervous and sensory system of oribatid mites, or about sensory integration or the neuronal bases of their behavior (Alberti, 1998; Alberti and Coons, 1999; Norton, 2007). Modern methods like Synchrotron X-ray microtomography (SRμCT) recently made it possible to investigate the organization and development of the nervous systems of oribatid mites (Figure 6e; (Hartmann et al., 2016)). We here provide the first genomic resource for the investigation of the photo- and chemosensory systems of Oribatida (Figure 6b-d). In addition, we give a conservative estimate of olfactory gene numbers based on automated detection algorithm which should be refined by manual curation in the future (Table 2; see'Materials & Methods'). For instance, manual curation of olfactory genes revealed additional chemosensory receptor gene models in the spider mite (Ngoc et al., 2016).
Horizontal gene transfer event sheds light on oribatid feeding biology

Horizontal gene transfer (HGT) is common among mites and other soil organisms (Grbić et al., 2011; Mayer et al., 2011; Faddeeva-Vakhrusheva et al., 2016; Wu et al., 2017; Dong et al., 2018; Wybouw et al., 2018). In some cases, genes that had been horizontally transferred now have pivotal biological functions. For instance, terpene and carotenoid biosynthesis genes in trombidiid and tetranychid mites show high homology with bacterial (terpene synthase) or fungal (carotenoid cyclase/synthase/desaturase) genes, suggesting horizontal gene transfer from microbial donors (Altincicek et al., 2012; Dong et al., 2018). At least the carotenoid biosynthesis genes in spider mites still code for functional enzymes and equip these phytophages with the ability to de novo synthesize carotenoids, which can induce diapause in these animals (Altincicek et al., 2012; Bryon et al., 2017).
Soil microarthropods like collembolans show numbers of horizontally transferred genes that are among the highest found in metazoan genomes, exceeded only by nematodes living in decaying organic matter (Crisp et al., 2015; Faddeeva-Vakhrusheva et al., 2016; Wu et al., 2017). Interestingly, many HGT genes found in collembolans are involved in carbohydrate metabolism and were especially enriched for enzyme families like glycoside hydrolases, carbohydrate esterases or glycosyltransferases (Faddeeva-Vakhrusheva et al., 2016; Wu et al., 2017). All three enzyme families are involved in the degradation of plant and fungal cell walls (Latgé, 2007; Gilbert, 2010). Hence, it has been hypothesized that cell-wall degrading enzymes acquired by HGT are beneficial for soil organisms as it allowed such animals to access important food source in a habitat that is highly biased towards polysaccharide-rich resources (Mitreva et al., 2009; Faddeeva-Vakhrusheva et al., 2016; Faddeeva-Vakhrusheva et al., 2017; Wu et al., 2017).
To assess the degree of HGT in Archegozetes we first used blobtools (v1.0) (Laetsch and Blaxter, 2017) to generate a GC proportion vs read coverage plot of our long-read genome assembly, in order to remove contigs of bacterial origin (Figure 7a; 438 contigs). After Hi-C scaffolding we removed another 94 scaffolds of bacterial origin amounting to a total of ~ 9 Mb of contamination. Of the remaining scaffolds, candidate HGTs were identified using the Alien Index (Flot et al., 2013; Thorpe et al., 2018), where HGTs are those genes with blast homology (bit score) closer to non-metazoan than metazoan sequences (supplementary Table S4). We further filtered these HGT candidates to remove those that overlapped predicted repeats by ≥ 50%, resulting in 748 genes. As HGT become integrated into the host genome, they begin to mirror features of the host genome, including changes in GC content and introduction of introns (Lawrence, 1997). Comparing the GC content of the HGT candidates showed two distinct peaks, one at 53.9% and the other at 34.2%, slightly higher than the remaining Archegozetes genes, GC content of 31.8% (Figure 7b). Of the 429 HGT genes that shared similar GC content to the host genome, 78.8% had at least one intron (Table S4). In a final step, we used the gene expression data (RNAseq) to filter the list of all putative HGT genes and only retained candidates that were expressed in any life stage of Archegozetes (n= 399 HGT genes).
The majority of HGT candidates were of bacterial origin (79.9%), followed by genes likely acquired from fungi (11.8%), while transfer from Archaea, plants, virus, and other sources was comparatively low (Figure 7d). This composition of HGT taxonomic origin is different from genes found in collembolans, which appear to have acquired more genes of fungal and protist origin (Faddeeva-Vakhrusheva et al., 2016; Faddeeva-Vakhrusheva et al., 2017; Wu et al., 2017). Subsequently, we performed an over-representation analysis of GO terms associated with these genes. We found an over-representation of genes with GO terms related to carbon-nitrogen ligase activity and hydrolase activity on glycosyl bonds (molecular function; Figure 7c) as well as carbohydrate metabolism (biological process; Figure 7c). This provides a first line of evidence that Archegozetes possess HGT related to plant- and fungal cell wall degradation similar to collembolans, yet mite and collembolan genes did not appear to be direct homologs. For instance, genes related to cell wall degrading enzymes in mites appear to be acquired by HGT from Streptomyces bacteria (Figure 7) while they are of fungal origin in collembolans (Faddeeva-Vakhrusheva et al., 2016; Faddeeva-Vakhrusheva et al., 2017; Wu et al., 2017).
As mentioned previously, oribatid mites are among the few Chelicerata that ingest solid food and are primary- and secondary decomposers feeding on dead plant material and fungi (Cohen, 1995; Norton, 2007; Shultz, 2007; Dunlop and Alberti, 2008; Heethoff and Norton, 2009; Maraun et al., 2011). It was argued for decades that the enzymes necessary to break down these polysaccharide-rich resources originate from the mite's gut microbes (Stefaniak, 1976; 1981; Smrž, 1992; Siepel and de Ruiter-Dijkman, 1993; Smrž, 2000; Smrž and Norton, 2004; Smrž and Čatská, 2010). Microbes might be mixed with the food in the ventriculus and digest it while passing through the alimentary tract as food boli enclosed in a peritrophic membrane (see Figure 7f for an example) (Stefaniak, 1976; 1981). However, screening the HGT candidate list for potential cell-wall degrading enzymes and mapping their overall and life-stage specific expression in Archegozetes using the RNAseq reads, revealed at least five HGT genes related to polysaccharide breakdown (Figure 7g). We found that specifically members of the glycoside hydrolases family 48 and cellulose-binding domain genes showed high expression in most life stages - the egg being an obvious exception (Figure 7g). Moreover, the majority of these genes were flanked by a predicted metazoan gene, suggesting host transcriptional regulation (Table S4).
In a last step we blasted the highly expressed HGT candidates (Figure 7g) against the non-redundant protein sequence database, aligned the sequences with the highest alignment score and performed a phylogenetic maximum likelihood analysis. For the highest expressed HGT related to cell-wall-degrading enzymes (glycoside hydrolases family 48 gene), we recovered that the Archegozetes sequences was well nested within a clade of GH 48 sequences from herbivores beetles (McKenna et al., 2019), which appear to be related to similar genes from various Streptomyces (Figure 7e) and we reconstructed similar phylogenies for other highly expressed HGT candidates (supplementary Figure S12). All the sequences of beetle glycoside hydrolases family 48 members (Figure 7e) were included in recent studies arguing for a convergent horizontal transfer of bacterial and fungal genes that enabled the digestion of lignocellulose from plant cell walls in herbivores beetles (McKenna et al., 2016; McKenna et al., 2019). They showed that phytophagous beetles likely acquired all genes of the GH 48 family from Actinobacteria (including Streptomyces) (McKenna et al., 2019) and our phylogenetic analysis (Figure 7e) revealed the same pattern as well as a highly similar tree topology (compare to Fig 3B in (McKenna et al., 2019)).
Overall, our findings indicate that genes encoding for enzymes in Archegozetes capable of degrading plant and fungal cell walls were likely horizontally transferred from bacteria (likely Streptomyces). Bacterial symbionts and commensal living in the mites' gut are still likely to contribute to the breakdown of food (Figure 7f). Yet, the high expression of genes encoding cell-wall degrading enzymes (Figure 7g) as well as the evolutionary analyses of such genes (Figure 7e) suggest that Archegozetes – and potentially many other oribatid mites – are able to exploit polysaccharide-rich resources like dead plant material or chitinous fungi without microbial aid. Enzymological and microscopical investigation of Archegozetes have suggested that certain digestive enzymes (chitinase and cellulase) are only active when the mites consume a particular type of food (e.g. algae, fungi or filter paper) (Smrž and Norton, 2004). These results were interpreted as evidence that these enzymes are directly derived from the consumed food source (Smrž and Norton, 2004). By contrast, we argue that this instead confirms our findings of HGT: upon consumption of food containing either chitin or cellulose, gene expression of polysaccharide-degrading enzymes starts, and proteins can readily be detected. Further enzymological studies have placed oribatid mites in feeding guilds based on carbohydrase activity and also found highly similar enzyme activity between samples of mites from different times and locations (Luxton, 1972; 1979; 1981; 1982; Siepel and de Ruiter-Dijkman, 1993). Future functional studies can disentangle the contribution of the host and microbes to cell wall digestion and novel metabolic roles of the HGTs identified here.
Biosynthesis of monoterpenes – a common chemical defense compound class across oribatid mite

Oribatid and astigmatid mites are characterized by a highly diverse spectrum of natural compounds that are produced by and stored in so-called oil glands (for an example see Figure 8a) (Raspotnig, 2009; Raspotnig et al., 2011; Heethoff et al., 2016). These paired glands are located in the opisthosoma (i.e., the posterior part of chelicerate arthropods, analogous to the abdomen of insects) and are composed of a single-cell layer invagination of the cuticle (Figure 8f). As previously mentioned, mites use chemicals produced by these glands to protect themselves against environmental antagonists (predators or microbes) or use them as pheromones (Shimano et al., 2002; Raspotnig, 2006; 2009; Heethoff et al., 2011a; Heethoff and Raspotnig, 2012; Brückner et al., 2015; Heethoff and Rall, 2015). The monoterpene aldehyde citral – a stereoisomeric mixture of geranial ((E)-3,7-dimethylocta-2,6-dienal) and neral ((Z)-3,7-dimethylocta-2,6-dienal) – and its derivatives are widely detected compounds in glandular secretions of oribatids and astigmatids (Sakata et al., 1995; Sakata, 1997; Kuwahara et al., 2001; Sakata and Norton, 2001; Sakata and Norton, 2003; Kuwahara, 2004; Raspotnig et al., 2004; Koller et al., 2012). These monoterpenes have been called ''astigmatid compounds'' (Sakata and Norton, 2001) as they characterize the biochemical evolutionary lineage of major oribatid mite taxa (Mixonomata and Desmonomata) and almost all investigated astigmatid mites (Alberti, 1984; Sakata, 1997; Sakata and Norton, 2001; Kuwahara, 2004; Raspotnig, 2009).
The chemical cocktail released by Archegozetes consists of a blend of 10 compounds (Figure 8a) including two terpenes (approx. 45%)– neral and neryl formate – six hydrocarbons (approx. 15%) and two aromatic compounds (approx. 40%) (Sakata and Norton, 2003; Brückner and Heethoff, 2017). The hydrocarbons likely serve as solvents, while the terpenes and aromatics are bioactive compounds used in chemical alarm and defense (Shimano et al., 2002; Sakata and Norton, 2003; Raspotnig, 2006; Heethoff et al., 2011a). Recently, it was shown that Archegozetes synthesizes the two aromatic compounds using a polyketide-like head-to-tail condensation of (poly)-β-carbonyls via a horizontally acquired putative polyketide synthase (Brückner et al., 2020). Studies in Astigmata found that the monoterpenes of these mites appeared to be made de novo from (poly)-β-carbonyls as well and one study identified a novel geraniol dehydrogenase (GeDH), unrelated to those of bacteria, in Carpoglyphus lactis (Morita et al., 2004; Noge et al., 2005; Noge et al., 2008). To learn about the biosynthesis of astigmatid compounds in Archegozetes and demonstrate the mite's applicability as research model for biochemical pathway evolution, we used the novel genomic resources presented in this study.
First, we delineated the basic biochemical reactions likely to happen in the Archegozetes gland through a stable-isotope labeling experiment. We supplemented the diet of the mite with food containing 25% heavy 13C6 D-glucose and 10% antibiotics (a combination of three different antibiotics was fed, because this mixture is able to eliminate nearly all qPCR and FISH detectable bacteria found on the food and in the alimentary tract (Brückner et al., 2020)). To examine the incorporation of heavy 13C6 D-glucose and its metabolic products into neral (Figure 8b) and neryl formate (Figure 8c), we compared selected fragment ions (M+ and M+-46, respectively) using single ion mass spectrometry. Both neral and neryl formate showed consistent enrichment in their M+ to [M+4]+ and [M-46]+ to [M-46+4]+-ion series, indicating that Archegozetes used glycolysis breakdown products of 13C6 D-glucose for the biosynthesis of their monoterpenes. We then used the OGS mapped to KEGG metabolic pathways (Kanehisa et al., 2007) to reconstruct the backbone synthesis of terpenes in Archegozetes (Figure 8d). We found mite genes, which suggest that Archegozetes synthesizes geranyl pyrophosphate (GPP) – the input substrate for further monoterpene synthesis – via the mevalonate pathway using the Mevalonate-5P to Isopentenyl-PP route (Figure 8d). The Mevalonate-5P pathway is used in most higher eukaryotes as compared to the Mevalonate-3P pathway in Archaea and the MEP/DOXP pathway in bacteria, some plants and apicomplexan protists (Trapp and Croteau, 2001; Eisenreich et al., 2004; Breitmaier, 2006; Degenhardt et al., 2009; Miziorko, 2011; Oldfield and Lin, 2012). This likely excludes any horizontal gene transfer of mevalonate pathway genes as Archegozetes uses enzymes similar to those of other animals.
The biosynthesis of monoterpenes not only depends on very widespread enzymes, but also requires more specific enzymes downstream of GPP (Trapp and Croteau, 2001; Breitmaier, 2006; Degenhardt et al., 2009). For instance, Carpoglyphus lactis expresses a unique geraniol dehydrogenase (GeDH) – catalyzing the oxidation of geraniol to geranial – different from all previously characterized geraniol-related and alcohol dehydrogenases (ADHs) of animals and plants (Noge et al., 2008). We used the functionally validated Carpoglyphus-GeDH (Noge et al., 2008), blasted its sequence against the Archegozetes OGS and found a homologous sequence. We used both mite sequences in an alignment with plant, fungal and bacterial GeDHs and animal ADHs and constructed a maximum likelihood phylogeny (Figure 8e). Similar to the previous analysis including only Carpoglyphus-GeDH, we found that the Al-GeDH represent a new class of geraniol dehydrogenases different from those in plants, fungi or bacteria and not nested within animal ADHs (Figure 8e). This is why we hypothesize that Al-GeDH is a novel expansion of the geraniol dehydrogenases gene family and has not been acquired by horizontal gene transfer, like other biosynthesis and digestive enzymes in Archegozetes (Figure 7; (Brückner et al., 2020)).
Based on our mass spectrometry data of stable isotopes and genomic analysis, we propose that the following biochemical pathway leading to monoterpenes is of oribatid mites (Figure 8f and g): geraniol is likely to be synthesized from GPP – the universal precursor of all monoterpenes – either enzymatically by a geraniol synthase (GES) or a diphosphate phosphatase (DPP), but possibly also endogenously by dephosphorylation of GPP (Oswald et al., 2007; Zhou et al., 2014; Liu et al., 2015; Beran et al., 2019). For Archegozetes, we could not find any GES or specific DPP in the OGS, thus geraniol might be formed from GPP via endogenous dephosphorylation, but further research is required to verify or falsify this hypothesis. Subsequently, geraniol is oxidized to geranial by the pervious described GeDH (Figure 8e) and readily isomerized to neral. Trace amounts of geranial have been found in Archegozetes and it is common among other oribatid and astigmatid mites, supporting this idea (Kuwahara, 2004; Raspotnig et al., 2004; Raspotnig et al., 2008; Koller et al., 2012). Also, there is no evidence that geraniol is converted into nerol, or that neral is formed directly via oxidation of nerol (Morita et al., 2004; Noge et al., 2005; Noge et al., 2008). The most parsimonious explanation for neryl formate synthesis would be an esterification of the corresponding terpene alcohol nerol. There is, however, no evidence of nerol in the traces of any oribatid or astigmatid mite species (Kuwahara, 2004; Raspotnig, 2009; Raspotnig et al., 2011). Aliphatic non-terpene formats in Astigmata are synthesized by dehomologation and generation of a one-carbon–shorter primary alcohol from an aldehyde via hydrolysis of formate in a biological Baeyer–Villiger oxidation catalyzed by a novel, uncharacterized enzyme (Shimizu et al., 2017). A similar reaction to synthesize terpene formates is unlikely, as the terpenoid backbone would be shortened by one-carbon and this does not happen in any possible scenario. The discovery of this Baeyer–Villiger oxidation mechanism, however, highlights the probability that there are many very unusual reactions that remain to be discovered in oribatid mites (Brückner et al., 2020).
Conclusion
The integrated genomic and transcriptomic resources presented here for Archegozetes longisetosus allowed a number of insights into the molecular evolution and basic biology of decomposer soil mites. Our analysis of an oribatid mite genome also provides the foundation for experimental studies building on the long history of Archegozetes as a chelicerate model organism, which now enters the molecular genetics era (Aoki, 1965; Palmer and Norton, 1992; Norton et al., 1993; Heethoff et al., 2013). This includes the study of biochemical pathways, biochemistry, neuroethological bases of food searching behavior, and environmental impacts on genomes of complex, clonal organisms.
Our evolutionary comparisons across the Chelicerata revealed interesting patterns of genome evolution and how horizontal gene transfer might have shaped the feeding mode of soil mites. We also showed how oribatid glandular biology and chemical ecology are reflected in the genome. The community of researchers studying the fundamental biology of oribatid, and other free-living, non-parasitic mites is growing. We think that providing these genomic and transcriptomic resources can foster a community effort to eventually allowing for basic molecular research on these mites.
Key priorities for a future community research effort include i) sequencing organ-specific transcriptomic data, ii) developing tools for genetic interrogation (RNAi or CRISPR/CAS9), iii) establishing reporter linages with germ-line stable modifications (e.g. GAL4/UAS misexpression systems), and iv) constructing an whole-animal single-cell RNAseq expression atlas. Please do not hesitate to contact the corresponding author if you want to start your own culture of Archegozetes. We will be happy to provide you with starter specimens for free and share rearing protocols with you.
Material and methods
Mite husbandry
The lineage'ran' (Heethoff et al., 2013) of the pantropical, parthenogenetic oribatid mite Archegozetes longisetosus was used in this study. Stock cultures were established in 2015 from an already existing line and fed with wheat grass (Triticum sp.) powder from Naturya. Cultures were maintained at 20-24 °C and 90% relative humidity. Sterilized water and 3-5 mg wheat grass were provided three times each week.
DNA extraction and Illumina sequencing
For the short-read library, DNA was extracted from ~200 mites that were taken from the stock culture, starved for 24 h to avoid possible contamination from food in the gut, subsequently washed with 1% SDS for 10 s. For extraction of living specimens, we used the Quick-DNA Miniprep Plus Kit (Zymo Research) according to the manufacturer's protocol. Amounts and quality of DNA were accessed with Qubit dsDNA HS Kit (Thermo Fisher) and with NanoDrop One (Thermo Fisher) with target OD 260/280 and OD 260/230 ratios of 1.8 and 2.0-2.2, respectively. Extracted DNA was shipped to Omega Bioservices (Norcross, GA, USA) on dry ice for library preparation and sequencing. DNA library preparation followed the KAPA HyperPrep Kit (Roche) protocol (150 bp insert size), and 200 million reads were sequenced as 150bp paired-end on a HighSeq4000 (Illumina) platform.
High-molecular weight DNA isolation and Nanopore sequencing
Genomic DNA was isolated from ~300-500 mites starved for 24 h using QIAGEN Blood & Cell Culture DNA Mini Kit. Briefly, mites were flash frozen in liquid nitrogen and homogenized with a pestle in 1 ml of buffer G2 supplemented with RNase A and Proteinase K at final concentrations of 200 ng/µl and 1 µg/µl, respectively. Lysates were incubated at 50C for 2 h, cleared by centrifugation at 5 krpm for 5 min at room temperature and applied to Genomic tip G/20 equilibrated with buffer QBT. Columns were washed with 4 ml of buffer QC and genomic DNA was eluted with 2 ml of buffer QF. DNA was precipitated with isopropanol, washed with 70% EtOH and resuspended in 50 µl of buffer EB. DNA was quantified with Qubit dsDNA HS Kit (Thermo Fisher) and the absence of contaminants was confirmed with NanoDrop One (Thermo Fisher) with target OD 260/280 and OD 260/230 ratios of 1.8 and 2.0-2.2, respectively. DNA integrity was assessed using Genomic DNA ScreenTape kit for TapeStation (Agilent Technologies).
Libraries for nanopore sequencing were prepared from 1 µg of genomic DNA using 1D Genomic DNA by Ligation Kit (Oxford Nanopore) following manufacturer's instructions. Briefly, unfragmented DNA was repaired and dA tailed with a combination of NEBNext FFPE Repair Mix (New England Biolabs) and NEBNext End repair/dA-tailing Module (New England Biolabs). DNA fragments were purified with Agencourt AMPure XP beads (Beckman Coulter) and Oxford Nanopore sequencing adapters were ligated using NEBNext Quick T4 DNA Ligase (New England Biolabs). Following AMPure XP bead cleanup, ~500 ng of the library was combined with 37.5 µL of SQB sequencing buffer and 25.5 µl of loading beads in the final volume of 75 µl and loaded on a MinION Spot-ON Flow Cell version R9.4 (Oxford Nanopore). Two flow cells were run on MinION device controlled by MinKNOW software version 3.1.13 for 48 hours each with local basecalling turned off generating 9.7 and 5.1 GB of sequence data. Post run basecalling was performed with Guppy Basecalling Software, version 3.4.5 (Oxford Nanopore). After filtering low quality reads (Q\textless7), the combined output of the two runs was 13.69 GB and 4.7 million reads.
Hi-C library preparation
A sequencing library was constructed from approximately 150 mg of mites, or ~1000 individuals, using the Proximo Hi-C kit following specific instructions for insects (Phase Genomics). Library quantity and quality was assessed using Qubit dsDNA High Sensitivity Assay (Thermo Fisher) and bioanalyzer (Aligent), respectively. Two hundred million 150bp paired-end reads were sequenced on a NextSeq 2000 platform (Illumina) by the Millard and Muriel Jacobs Genetics and Genomics Laboratory at California Institute of Technology.
Genome assembly and contamination filtering
Read quality was assessed using FastQC v0.11.8 (Andrews, 2010). Illumina adapters, low-quality nucleotide bases (phred score below 15) from the 3′ and 5′ ends and reads shorter than 50 bp were removed using cutadapt v1.18 (Martin, 2011). From the filtered reads, in silico genome size estimates were calculated using k-mer based tools kmergenie v.1.7048 (Chikhi and Medvedev, 2014), GenomeScope v1.0 (Vurture et al., 2017), and findGSE v0.1.0 R package (Sun et al., 2018). The latter two required a k-mer histogram computed by jellyfish v2.2.10 (Marçais and Kingsford, 2011) with k-mer size of 21. The long-read genome was assembled using 4.7 million reads from two MinION runs (60x coverage) using Canu v1.8 with default settings and setting the expected genome size to 200 Mb (Koren et al., 2017). To improve assembly quality, paired end Illumina reads were mapped to the genome with BWA aligner (Li and Durbin, 2009) using BWA-MEM algorithm and polished with Pilon v. 1.23 with'—changes' and'–fix all' options (Walker et al., 2014). Assembled contigs identified as bacterial and fungal contaminants based on divergent GC content from most Archegozetes contigs, high coverage and blast homology to the nt database (downloaded February 2019, Evalue 1e-25) were removed using Blobtools v1.0 (Laetsch and Blaxter, 2017).
Duplicate contigs, or haplotigs, of the long-read assembly were removed using purge_haplotigs v.1.1 (Roach et al., 2018). The short-read data was mapped to the long-read assembly using minimap2 (Li, 2018) and then filtered based on read coverage (parameters: -l 25 -m 230 -h 500). The Hi-C sequencing data was then mapped to the purged assembly with BWA (Li and Durbin, 2009). The Hi-C contact map was generated and corrected from the mapped reads using hicexplorer (Ramírez et al., 2018) with a bin size of 10000 and z-score thresholds of -1.5 and 3 for correction. The contact map was then used by HiCAssembler (Renschler et al., 2019) along with the purged assembly for scaffolding (parameters: –min_scaffold_length 300000 –bin_size 20000 –num_iterations –3 split_positions_file split.bed). The utility tool plotScaffoldInteractive was used to identify coordinates of misassemblies to manually split in the assembly process (split.bed).
Identification, classification, and masking of repetitive element
Repetitive elements in the genome Archegozetes were identified using a species-specific library generated with RepeatModeler v 1.0.11 (Smit and Hubley, 2008; Bao et al., 2015) and MITE tracker (Crescente et al., 2018) and annotated by RepeatClassifier, a utility of the RepeatModeler software that uses the RepBase database (version Dfam_Consensus-20181026). Unclassified repeat families from both programs were run through CENSOR v 4.2.29 (Kohany et al., 2006) executable censor.ncbi against the invertebrate library v 19.03 to provide further annotation. Predicted repeats were removed if they had significant blast homology (E-value 1e-5) to genuine proteins in the NCBI nr database and/or a local database of arthropod genomes (Drosophila melanogaster, Tribolium castaneum, Tetranychus urticae, Leptotrombidium deliense, Dinothrombium tinctorium, Sarcoptes scabiei, Euroglyphus maynei, Galendromus occidentalis, Dermatophagoides pteronyssinus). Unclassified repeats with blast homology to known TEs were retained whereas those with no blast homology were removed (Petersen et al., 2019). The remaining repeat families were combined with the Arthropoda sequences in RepBase and clustered using vsearch v 2.7.1 (–iddef 1 –id 0.8 –strand both; (Rognes et al., 2016)). The filtered repeat library was used to soft mask the A. longisetosus Hi-C assembly using RepeatMasker v 4.07 (Smit et al., 1996-2010). A summary of the masked repeat content was generated using the ''buildSummary.pl'' script, the Kimura sequence divergence calculated using the ''calcDivergenceFromAlign.pl'' script and the repeat landscape visualized using the ''createRepeatLandscape.pl'' script, all utilities of RepeatMasker.
Gene prediction and annotation
Both ab inito and reference-based tools were used for gene prediction of the long-read assembly using modified steps of the funannotate pipeline (Palmer and Stajich, 2017). The ab inito tool GeneMark-ES v4.33 (Ter-Hovhannisyan et al., 2008) was used along with reference based tools BRAKER v2.1.2 (Bruna et al., 2020) using RNAseq reads discussed below and PASA v 2.3.3 (Haas et al., 2008) using genome-guided transcriptome assembly from Trinity described below. Lastly, Tetranychus urticae gene models from the NCBI database (GCF_000239435.1) were aligned to the contigs using GeMoMa (Keilwagen et al., 2019). All gene predictions were combined in EVidenceModeler (Haas et al., 2008) with the following weights: GeMoMa =1, PASA = 10, other BRAKER = 1, and GeneMark = 1. Predicted tRNAs using tRNAscan-SE v 2.0.3 (Chan and Lowe, 2019) were combined with the gene predictions in the final gene feature format (GFF) file and filtered for overlap using bedtools (Quinlan and Hall, 2010) intersect tool (Quinlan and Hall, 2010). After constructing the Hi-C assembly, gene predictions above were assigned to the new coordinates using a combination of Liftoff (Shumate and Salzberg, 2021) and GeMoMa (Keilwagen et al., 2019), both with default settings. The new assembly was also reanalyzed with PASA using the transcriptome as described above to recover incomplete open-reading frames from the lift over procedure. The gene predictions were combined with EVidenceModeler with the following weights: GeMoMa= 5, Liftoff = 5, and PASA = 10.
The predicted genes were searched against the NCBI nr (February 2019) (Pruitt et al., 2005), SwissProt (February 2019) (Bairoch and Apweiler, 2000), a custom-made Chelicerata database including genomes of Tetranychus urticae, Leptotrombidium deliense, Dinothrombium tinctorium, Sarcoptes scabiei, Euroglyphus maynei, Galendromus occidentalis, Metaseiulus occidentalis, Dermatophagoides pteronyssinus, Trichonephila clavipes, Stegodyphus mimosarum, Centruroides sculpturatus, Ixodes scapularis and Parasteatoda tepidariorum (all downloaded Feb 2019), PFAM (v 32, August 2018) (Bateman et al., 2004), merops (v 12, October 2017) (Rawlings et al., 2010) and CAZY (v 7, August 2018) (Cantarel et al., 2009) databases. The results of the hmm-based (Eddy, 2011) PFAM and CAZY searches were filtered using cath-tools v 0.16.2 (https://cath-tools.readthedocs.io/en/ ![]() ; E-value 1e-5) and the blast-based searches were filtered by the top hit (E-value 1e-5 threshold). Predicted genes were also assigned to orthologous groups using eggNOG-mapper (Huerta-Cepas et al., 2017). Gene annotation was prioritized by the SwissProt hit if the E-value < 1e-10 followed by NCBI annotation, the custom Chelicerata database and if no homology was recovered, then the gene was annotated as, ''hypothetical protein''.
; E-value 1e-5) and the blast-based searches were filtered by the top hit (E-value 1e-5 threshold). Predicted genes were also assigned to orthologous groups using eggNOG-mapper (Huerta-Cepas et al., 2017). Gene annotation was prioritized by the SwissProt hit if the E-value < 1e-10 followed by NCBI annotation, the custom Chelicerata database and if no homology was recovered, then the gene was annotated as, ''hypothetical protein''.
Analysis of the official gene set (OGS)
To allow the OGS to be used as resources for functional studies, we assigned functional categories based on Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000; Consortium, 2004). GO terms for the respective genes models of the OGS were assigned based on the gene id with highest homology from the SwissProt database or NCBI nr database (Bairoch and Apweiler, 2000; Pruitt et al., 2005). A custom database of GO terms was created with makeOrgPackage function in the R package AnnotationForge v1.26.0 (Carlson and Pagès, 2019). Over-representation analysis of GO terms was tested using the enrichGO function in the R package clusterProfiler v3.12.0 (Yu et al., 2012) with a hypergeometric distribution and a Fisher's Exact test. P-values were adjusted for multiple comparisons using false discovery rate correction (Benjamini and Hochberg, 1995). Resulting enriched GO terms were processed with GO slim (Consortium, 2019) and the final list of over represented GO terms was used to plot the number of genes in a respective category.
KEGG orthology terms were assigned from single-directional best hit BLAST searches of each gene model on the KEGG Automatic Annotation Server (Moriya et al., 2007). Additionally, we ran GhostKOALA (Kanehisa et al., 2016) (GHOSTX searches for KEGG Orthology And Links Annotation) to obtain KEGG orthology terms. Compared to conventional BLAST searches, GhostKOALA is about 100 times more efficient than BLAST to remote homologs by using suffix arrays (Suzuki et al., 2014).
Orthology and phylogenomic analyses
Orthologs of A. longisetosus, other species within Acari, Chelicerata and the fruit fly Drosophila were identified using OrthoFinder v 2.3.3 (-M msa –A mafft –T fasttree; (Emms and Kelly, 2015)). Prior to running OrthoFinder, isoform variants were removed from the gene predictions using CD-Hit (Fu et al., 2012). Trees of orthogroups with at least 80% of taxa present (n= 4,553) were constructed using fasttree v 2.1.10 (Price et al., 2010), trimmed with TrimAl v 1.4.1 (-keepheader -fasta –gappyout; (Capella-Gutiérrez et al., 2009)) and paralogs pruned using phylotreepruner v 1.0 (min_number_of_taxa =18, bootstrap_cutoff= 0.7, longest sequence for a given orthogroup=u; (Kocot et al., 2013)). Alignments shorter than 100 amino acids were removed, leaving 1,121 orthogroups.
For the maximum likelihood analysis, the trimmed and pruned alignments were concatenated into a supermatrix using FasConCat v1.04 (Kück and Meusemann, 2010) composed of 377,532 amino acids and the best substitution models determined using PartitionFinder v 2.1.1 (Lanfear et al., 2016). The maximum likelihood consensus phylogeny from the supermatrix and partition scheme was constructed using IQ-tree and 1,000 ultrafast bootstrap replicates (Nguyen et al., 2015). For the coalescence species tree reconstruction, gene trees were generated using IQ-tree v 1.6.12 on the trimmed alignments of the 1,121 filtered orthogroups and processed using ASTRAL v 5.6.3 (Zhang et al., 2018). Branch lengths are presented in coalescent units (differences in the 1,121 gene trees) and the node values reflect the local posterior probabilities.
RNA sequencing and transcriptome assembly
For RNA extraction, about 200 mites of all life stages were taken from stock culture and subsequently washed with 1% SDS for 10 s. RNA was extracted from living specimens using the Quick-RNA MiniPrep Kit (Zymo Research) according to the manufacturer's protocol. Quantity and quality of RNA were accessed using a Qubit fluorometer and NanoDrop One (Thermo Fisher Scientific), respectively.
Extracted RNA was shipped to Omega Bioservices (Norcross, GA, USA) on dry ice for library preparation and sequencing. Whole animal RNA was used for poly-A selection, cDNA synthesis and library preparation following the Illumina TruSeq mRNA Stranded Kit protocol. The library was sequenced with 100 million 150 bp paired-end on a HighSeq4000 platform. For the genome-guided assembly of the transcriptome a bam-file was created from the long-read genome using STAR (Dobin et al., 2013). RNAseq reads were in silico normalized and subsequently used together with the bam-file to assemble the transcripts using Trinity v2.8.4 (Grabherr et al., 2011; Haas et al., 2013), yielding an assembly with a total length of 162.8 Mb, an N50= 2994 bp and a BUSCO score (Simão et al., 2015) of C:96.3% [S:36.5%,D:59.8%], F:1.3%, M:2.4%.
Life-stage specific RNAseq
For life-stage specific RNAseq, we collected 15 specimens per life stage from the stock culture that were split into three replicates of five individuals. Whole animals (for all stages but eggs) were flash frozen in 50 µl TRIzol using a mixture of dry ice and ethanol (100%) and stored at −80°. RNA was extracted using a combination of the TRIzol RNA isolation protocol (Life Technologies) and RNeasy Mini Kit (Qiagen) (Kitchen et al., 2015). The TRIzol protocol was used for initial steps up to and including the chloroform extraction. Following tissue homogenization, an additional centrifugation step was performed at 12,000 × g for 10 min to remove tissue debris. After the chloroform extraction, the aqueous layer was combined with an equal volume of ethanol and the RNeasy Mini Kit was used to perform washes following the manufacturer's protocol. Eggs were crushed using pipette tips and directly stored in a mixture of cell lysis buffer and murine RNase Inhibitor (New England Biolab).
We used the NEBNext® Single Cell/Low Input RNA Library Prep Kit for Illumina® together with NEBNext® Multiplex Oligos for Illumina® (New England Biolab) for library preparation, including reverse transcription of poly(A) RNA, amplification full-length cDNA, fragmentation, ligation and final library amplification according to the manufacturer's protocol. We performed cDNA amplification for 16 (18 for egg samples) PCR cycles and final library amplification 8 PCR cycles. In total, we constructed 18 libraries (three for each life stage). The quality and concentration of the resulting libraries were assessed using the Qubit High Sensitivity dsDNA kit (Thermo Scientific) and Agilent Bioanalyzer High Sensitivity DNA assay. Libraries were sequenced on an Illumina HiSeq2500 platform (single-end with read lengths of 50 bp) with ~18 million reads per library.
Illumina sequencing reads were pseudoaligned to the bulk transcriptome and quantified (100 bootstrap samples) with kallisto 0.46.0 (Bray et al., 2016) using default options for single-end reads. Fragment length sizes were extracted from the Agilent Bioanalyzer runs. For life-stage specific differential expression analysis, kallisto quantified RNAseq data was processes with sleuth 0.30.0 (Pimentel et al., 2017) using Likelihood Ratio tests in R 3.6.1 (RCoreTeam, 2019). The average transcripts per million (tpm) values for each target transcript were extracted from the sleuth object (see R script) and used with the Heatmapper tool (Babicki et al., 2016) to produce an unclustered heatmap showing relative expression levels. UpSetR (Conway et al., 2017) was used to compare the number of unique and shared expressed genes across life stages.
Identification of horizontal gene transfer events
To detect HGTs, we used the published tool ''.Lateral_gene_transfer_predictor.py'' (Thorpe et al., 2018) to calculate the Alien Index described by (Gladyshev et al., 2008) and (Flot et al., 2013). All predicted genes were compared to the NCBI nr database as previously described (Thorpe et al., 2018). Results to Arthropoda (tax id 6656) were ignored in the downstream calculations. The HGT candidates were filtered for contamination identified by both Blobtools (Laetsch and Blaxter, 2017) and the Alien Index (AI < 30 and \textgreater70% percent identity to a non-metazon sequence). The candidates were further filtered for < 50% overlap with predicted repeats using the bedtools intersect tool with the RepeatMasker gff file and expression from any developmental stage. Introns were scored manually from visualization in IGV genome browser (Robinson et al., 2011) and GC content for all predicted genes was calculated using the bedtools nuc tool.
Analysis of chemosensory and photoreceptor gene families
The search and analysis chemosensory genes largely followed the procedure outlined by Dong et al. (Dong et al., 2018) with slight modifications. First, the Archegozetes official gene set (OGS) was searched using BLASTP (E-value, \textless1 × 10−3) against the following queries for the different chemosensory gene families. The OGS was queried against i) D. melanogaster, D. mojavensis, Anopheles gambiae, Bombyx mori, T. castaneum, Apis mellifera, Pediculus humanus humanus, and Acyrthosiphon pisum odorant binding proteins (OBPs) (Vieira and Rozas, 2011); ii) D. melanogaster, D. mojavensis, A. gambiae, B. mori, T. castaneum, A. mellifera, P. humanus humanus, A. pisum, I. scapularis, and Daphnia pulex small chemosensory proteins (CSP) (Niimura and Nei, 2005; Robertson and Wanner, 2006; Vieira and Rozas, 2011); iii) D. melanogaster and A. mellifera odorant receptors (Niimura and Nei, 2005; Robertson and Wanner, 2006); iv) D. melanogaster, A. mellifera, I. scapularis, T. urticae, T. mercedesae, and M. occidentalis gustatory receptors (GRs) (Robertson et al., 2003; Robertson and Wanner, 2006; Gulia-Nuss et al., 2016; Hoy et al., 2016; Ngoc et al., 2016; Dong et al., 2017); v) a comprehensive list of iGluRs and IRs across vertebrates and invertebrates (Croset et al., 2010), as well as those identified in the T. mercedesae, D. tinctorium and L. deliense genome projects (Dong et al., 2017; Dong et al., 2018). Second, all candidate Archegozetes sequences were reciprocally blasted (BLASTP, E-value \textless1 × 10−3) against the NCBI database (Pruitt et al., 2005) and all sequences that did not hit one of the respective receptors or transmembrane proteins were removed from the list. Third, for phylogenetic analysis of IRs and GRs from Archegozetes were aligned with IRs from D. melanogaster, T. urticae, D. tinctorium and L. deliense and GRs from iv) D. melanogaster, T. mercedesae, I. scapularis, and M. occidentalis, respectively, using MAFFT (v 7.012b) with default settings (Katoh and Standley, 2013). Poorly aligned and variable terminal regions, as well as several internal regions of highly variable sequences were excluded from the phylogenetic analysis. Fourth, maximum likelihood trees were constructed with the IQ-TREE pipeline (v 1.6.12) with automated model selection using 1,000 ultrafast bootstrap runs (Nguyen et al., 2015).
Reference opsin genes and opsin-like sequences were obtained from Dong et al. (Dong et al., 2018) and used to query the Archegozetes OGS using BLASTP (E-value, \textless1 × 10−5). Subsequently, candidates sequenced were reciprocally blasted against NCBI using the same settings and only retained if they hit an opsin or opsin-like gene. The Archegozetes candidates were aligned with the query sequence list using MAFFT (v 7.012b) with default settings (Katoh and Standley, 2013). This opsin gene alignment phylogenetically analyzed using the IQ-TREE pipeline (v 1.6.12) with automated model selection and 1,000 ultrafast bootstrap runs (Nguyen et al., 2015).
Gene family phylogenies
We used the following workflow to analyses genes related to Figure 5 (hox and developmental genes), Figure 7 (cell wall-degrading enzyme encoding genes) and Figure 8 (alcohol and geraniol dehydrogenases genes). Generally, protein orthologs were retrieved from NCBI (Pruitt et al., 2005), and aligned using MUSCLE (Edgar, 2004) or MAFFT (v 7.012b) (Katoh and Standley, 2013) and ends were manually inspected and trimmed. The resulting final protein sequence alignments used to construct a maximum likelihood (ML) phylogenetic tree with either i) PhyML with Smart Model Selection (Guindon et al., 2010; Lefort et al., 2017) or ii) the IQ-TREE pipeline with automated model selection (Nguyen et al., 2015). The ML trees were constructed using either 1,000 ultrafast bootstrap runs (IQ-TREE) or approximate-likelihood ratio test (PhyML) was used to assess node support.
Feeding experiments with labelled precursors and chemical analysis (GC/MS)
Stable isotope incorporation experiments were carried out as previously described (Brückner et al., 2020). Briefly, mites were fed with wheat grass containing a 10% (w/w) mixture of three antibiotics (amoxicillin, streptomycin and tetracycline) and additionally, we added 25% (w/w) of the stable isotope-labelled precursors [13C6] D-glucose (Cambridge Isotope Laboratories, Inc.) as well as a control with untreated wheat grass. Cultures were maintained for one generation and glands of adult specimens were extracted one week after eclosion by submersing groups of 15 individuals in 50 µl hexane for 5 min, which is a well-established method to obtain oil gland compounds from mites (Raspotnig et al., 2008; Brückner and Heethoff, 2016; 2017; Brückner et al., 2017b).
Crude hexane extracts (2-5 µl) were analysed with a GCMS-QP2020 gas chromatography – mass spectrometry (GCMS) system from Shimadzu equipped with a ZB-5MS capillary column (0.25 mm x 30m, 0.25 µm film thickness) from Phenomenex. Helium was used a carrier gas with a flow rate of 2.14 ml/min, with splitless injection and a temperature ramp was set to increase from 50 °C (5 min) to 210 °C at a rate of 6 °C/min, followed by 35 °C/min up to 320 °C (for 5 min). Electron ionization mass spectra were recorded at 70 eV and characteristic fragment ions were monitored in single ion mode. The temperatures of the ion source and transfer line were 230 °C and 320 °C, respectively.
Abbreviations
13C= stable isotope of carbon with the isotopic mass 13; 15N= stable isotope of nitrogen with the isotopic mass 15; abd-A = abdominal A; Abd-B= Abdominal-B; ADHs= alcohol dehydrogenases; ADU= adult; BLAST= basic local alignment search tool; bp= base pairs; BUSCO: C= complete genes, S= single copy genes, D= doublet genes, F= fragmented genes, M= missing genes; BUSCO= Benchmarking Universal Single-Copy Orthologs; Ch= chelicera; CRISPR/CAS9= clustered regularly interspaced short palindromic repeats/ CRISPR associated protein 9; CSP= chemosensory protein; dac= dachshund; DAPI= 4′,6-diamidino-2-phenylindole; DEU= deutonymph; Dll= Distal-less; EGG= egg; EtOH= ethanol; exd= extradenticle; eya= eyes absent; FISH= fluorescence in situ hybridization; Gb= gigabase pairs, GeDH= geraniol dehydrogenase; GES= geraniol synthase; GFF= general feature format; GO= Gene Ontology; GPP= geranyl pyrophosphate; GR= gustatory receptor; HCN= hydrogen cyanide; HGT= horizontal gene transfer; Hi-C= all-versus-all chromosome conformation capture sequencing; hth= homothorax; iGluR= ionotropic glutamate receptors; IR= ionotropic receptor; kb= kilobase pairs, KEGG= Kyoto Encyclopedia of Genes and Genomes; KOALA= KEGG Orthology And Links Annotation; L1-3= walking legs 1-3; LAR= larva; LTR= Long Terminal Repeat; Mb = megabase pairs, MEP/DOXP pathway= 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate pathway; NCBI= National Center for Biotechnology Information; NMDA= N-methyl-D-aspartic acid; NMDS= non-metric multidimension scaling; OBP= odorant binding protein; OD= optical density; OGS= official gene set; OR= odorant receptor; PCR= polymerase chain reaction; PFAM= Protein Families; Pp= pedipalp; PRO= protonymph; qPCR= quantitative polymerase chain reaction; ran= Roy A. Norton; RNAi= RNA interference; RNAseq= RNA sequencing; SEM= scanning electron microscopy;so= six1/sine oculis; SRμCT= Synchrotron X-ray microtomography; TEM= transmission electron microscopy; TEs= transposable elements; tpm= transcripts per million; TRI= tritonymph; Ubx= Ultrabithorax
Declarations
Acknowledgment
We thank Joe Parker for making his laboratory space and resources available to us. Michael Heethoff, Sebastian Schemlzle, Benjamin Weiss and Martin Kaltenpoth graciously allowed us to use some of their unpublished images. Roy A. Norton provided invaluable comments to the manuscript and collected the first specimens of Archegozetes longisetosus giving rise to the current laboratory strain. AB thanks Joe Aragon for his help with the figure design.
Ethics statement
There are no legal restrictions on working with mites.
Authors contributions
AB had the initial idea for the study; AB, AAB, PB and SAK designed research; IAA performed long-read and Hi-C sequencing and assembled the long-read genome; AB performed all other experimental work; AAB analyzed hox and life-stage specific expression data; AB analyzed chemical data; SAK and AB performed bioinformatic analyses; AB wrote the first draft of the manuscript with input from AAB, PB, and SAK; SAK revised the manuscript. All authors gave final approval for publication.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Funding
This work was supported by a grant from the Caltech Center for Environmental Microbial Interactions (CEMI) to AB. AB was Simons Fellow of the Life Sciences Research Foundation (LSRF).
Data availability
Genomic and transcriptomic data generated for his project can be found on NCBI under the accession numbers PRJNA683935 and PRJNA683999. All other data that support the findings of this study have been deposited at https://doi.org/10.22002/D1.1877 ![]() (Brückner, 2021). Mite specimens to start an own laboratory culture can be requested from the corresponding author.
(Brückner, 2021). Mite specimens to start an own laboratory culture can be requested from the corresponding author.
acarologia_4528_Supplementary-figures.pdf
acarologia_4528_Supplementary-tables.xlsx
References
- Alberti G. 1984. The contribution of comparative spermatology to problems of acarine systematics. Acarology VI: 479-490.
- Alberti G. 1991. Spermatology in the Acari: systematic and functional implications. In: Schuster R., Murphy P.W., (Eds). The Acari - Reproduction, Development and Life-History Strategies. London: Chapman & Hall. p. 77-105. https://doi.org/10.1007/978-94-011-3102-5_6
- Alberti G. 1998. Fine structure of receptor organs in oribatid mites (Acari). In: Ebermann E., (Ed). Arthropod biology: Contributions to morphology, ecology and systematics. Wien: Austrian Academy of Sciences Press p. 27-77.
- Alberti G., Coons L.B. 1999. Acari-Mites. New York: Wiley. pp. 1265.
- Alberti G., Michalik P. 2004. Feinstrukturelle Aspekte der Fortpflanzungssysteme von Spinnentieren (Arachnida). Denisia, 12: 1-62.
- Alberti G., Moreno-Twose A.I. 2012. Fine structure of the primary eyes in Heterochthonius gibbus (Oribatida, Heterochthoniidae) with some general remarks on photosensitive structures in oribatid and other actinotrichid mites. Soil Org, 84: 391-408.
- Altincicek B., Kovacs J.L., Gerardo N.M. 2012. Horizontally transferred fungal carotenoid genes in the two-spotted spider mite Tetranychus urticae. Biol Lett, 8: 253-257. https://doi.org/10.1098/rsbl.2011.0704
- Andrews S. 2010. FastQC: A quality control tool for high throughput sequence data. Ref Source: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
 .
. - Aoki J. 1965. Oribatiden (Acarina) Thailands. I. Nat Life Southeast Asia, 4: 129-193.
- Arkhipova I., Meselson M. 2000. Transposable elements in sexual and ancient asexual taxa. PNAS, 97: 14473-14477. https://doi.org/10.1073/pnas.97.26.14473
- Babicki S., Arndt D., Marcu A., Liang Y., Grant J.R., Maciejewski A., Wishart D.S. 2016. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res, 44: W147-W153. https://doi.org/10.1093/nar/gkw419
- Bairoch A., Apweiler R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res, 28: 45-48. https://doi.org/10.1093/nar/28.1.45
- Ballesteros J.A., Sharma P.P. 2019. A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error. Syst Biol, 68: 896-917. https://doi.org/10.1093/sysbio/syz011
- Bao W., Kojima K.K., Kohany O. 2015. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA, 6: 11. https://doi.org/10.1186/s13100-015-0041-9
- Barnett A.A., Thomas R.H. 2012. The delineation of the fourth walking leg segment is temporally linked to posterior segmentation in the mite Archegozetes longisetosus (Acari: Oribatida, Trhypochthoniidae). Evol Dev, 14: 383-92. https://doi.org/10.1111/j.1525-142X.2012.00556.x
- Barnett A.A., Thomas R.H. 2013a. The expression of limb gap genes in the mite Archegozetes longisetosus reveals differential patterning mechanisms in chelicerates. Evol Dev, 15: 280-92. https://doi.org/10.1111/ede.12038
- Barnett A.A., Thomas R.H. 2013b. Posterior Hox gene reduction in an arthropod: Ultrabithorax and Abdominal-B are expressed in a single segment in the mite Archegozetes longisetosus. EvoDevo, 4: 23. https://doi.org/10.1186/2041-9139-4-23
- Barnett A.A., Thomas R.H. 2018. Early segmentation in the mite Archegozetes longisetosus reveals conserved and derived aspects of chelicerate development. Dev Genes Evol, 228: 213-217. https://doi.org/10.1007/s00427-018-0615-x
- Barrero R.A., Guerrero F.D., Black M., McCooke J., Chapman B., Schilkey F., de Leon A.A.P., Miller R.J., Bruns S., Dobry J. 2017. Gene-enriched draft genome of the cattle tick Rhipicephalus microplus: assembly by the hybrid Pacific Biosciences/Illumina approach enabled analysis of the highly repetitive genome. Int J Parasitol, 47: 569-583. https://doi.org/10.1016/j.ijpara.2017.03.007
- Barton N.H. 2010. Mutation and the evolution of recombination. Philos Trans R Soc Lond B Biol Sci, 365: 1281-1294. https://doi.org/10.1098/rstb.2009.0320
- Bast J., Schaefer I., Schwander T., Maraun M., Scheu S., Kraaijeveld K. 2016. No accumulation of transposable elements in asexual arthropods. Mol Biol Evol, 33: 697-706. https://doi.org/10.1093/molbev/msv261
- Bateman A., Coin L., Durbin R., Finn R.D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E.L. 2004. The Pfam protein families database. Nucleic Acids Res, 32: D138-D141. https://doi.org/10.1093/nar/gkh121
- Benjamini Y., Hochberg Y. 1995. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B, 57: 289-300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
- Bensoussan N., Santamaria M.E., Zhurov V., Diaz I., Grbić M., Grbić V. 2016. Plant-herbivore interaction: dissection of the cellular pattern of Tetranychus urticae feeding on the host plant. Front Plant Sci, 7: 1105. https://doi.org/10.3389/fpls.2016.01105
- Benton R., Vannice K.S., Gomez-Diaz C., Vosshall L.B. 2009. Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell, 136: 149-162. https://doi.org/10.1016/j.cell.2008.12.001
- Beran F., Köllner T.G., Gershenzon J., Tholl D. 2019. Chemical convergence between plants and insects: biosynthetic origins and functions of common secondary metabolites. New Phytol. https://doi.org/10.1111/nph.15718
- Bergmann P., Laumann M., Norton R.A., Heethoff M. 2018. Cytological evidence for automictic thelytoky in parthenogenetic oribatid mites (Acari, Oribatida): Synaptonemal complexes confirm meiosis in Archegozetes longisetosus. Acarologia, 58: 342-356. https://doi.org/10.24349/acarologia/20184246
- Bourque G., Burns K.H., Gehring M., Gorbunova V., Seluanov A., Hammell M., Imbeault M., Izsvák Z., Levin H.L., Macfarlan T.S. 2018. Ten things you should know about transposable elements. Genome Biol, 19: 1-12. https://doi.org/10.1186/s13059-018-1577-z
- Brandt A., Van P.T., Bluhm C., Anselmetti Y., Dumas Z., Figuet E., François C.M., Galtier N., Heimburger B., Jaron K.S. 2021. Haplotype divergence supports long-term asexuality in the oribatid mite Oppiella nova. Proceedings of the National Academy of Sciences, 118. https://doi.org/10.1073/pnas.2101485118
- Bray N.L., Pimentel H., Melsted P., Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotech, 34: 525-527. https://doi.org/10.1038/nbt.3519
- Breitmaier E. 2006. Terpenes: flavors, fragrances, pharmaca, pheromones. Weinheim: John Wiley & Sons. https://doi.org/10.1002/9783527609949
- Brückner A. 2021. Data related to "The Archegozetes longisetosus genome project". CaltechDATA. In: CaltechDATA, (Ed). 1.0 ed. Pasadena: CaltechDATA. https://doi.org/10.22002/D1.1876
- Brückner A., Heethoff M. 2016. Scent of a mite: origin and chemical characterization of the lemon-like flavor of mite-ripened cheeses. Exp Appl Acarol, 69: 249-61. https://doi.org/10.1007/s10493-016-0040-7
- Brückner A., Heethoff M. 2017. The ontogeny of oil gland chemistry in the oribatid mite Archegozetes longisetosus Aoki (Oribatida, Trhypochthoniidae). Int J Acarol, 43: 337-342. https://doi.org/10.1080/01647954.2017.1321042
- Brückner A., Heethoff M. 2018. Nutritional effects on chemical defense alter predator-prey dynamics. Chemoecology, 28: 1-9. https://doi.org/10.1007/s00049-018-0253-9
- Brückner A., Hilpert A., Heethoff M. 2017a. Biomarker function and nutritional stoichiometry of neutral lipid fatty acids and amino acids in oribatid mites. Soil Biol Biochem, 115: 35-43. https://doi.org/10.1016/j.soilbio.2017.07.020
- Brückner A., Kaltenpoth M., Heethoff M. 2020. De novo biosynthesis of simple aromatic compounds by an arthropod (Archegozetes longisetosus). Proc R Soc Lond Biol, 287: 20201429. https://doi.org/10.1098/rspb.2020.1429
- Brückner A., Parker J. 2020. Molecular evolution of gland cell types and chemical interactions in animals. J Exp Biol, 223. https://doi.org/10.1242/jeb.211938
- Brückner A., Raspotnig G., Wehner K., Meusinger R., Norton R.A., Heethoff M. 2017b. Storage and release of hydrogen cyanide in a chelicerate (Oribatula tibialis). PNAS, 114: 3469-3472. https://doi.org/10.1073/pnas.1618327114
- Brückner A., Schuster R., Smit T., Heethoff M. 2018a. Imprinted or innated food preferences in the model mite Archegozetes longisetosus (Actinotrichida, Oribatida, Trhypochthoniidae). Soil Org, 90: 23-26.
- Brückner A., Schuster R., Smit T., Pollierer M.M., Schäffler I., Heethoff M. 2018b. Track the snack - Olfactory cues shape foraging behaviour of decomposing soil mites (Oribatida). Pedobiologia, 66. https://doi.org/10.1016/j.pedobi.2017.10.004
- Brückner A., Schuster R., Wehner K., Heethoff M. 2018c. Effects of nutritional quality on the reproductive biology of Archegozetes longisetosus (Actinotrichida, Oribatida, Trhypochthoniidae) Soil Org, 90: 1-12.
- Brückner A., Stabentheiner E., Leis H.J., Raspotnig G. 2015. Chemical basis of unwettability in Liacaridae (Acari, Oribatida): specific variations of a cuticular acid/ester-based system. Exp Appl Acarol, 66: 313-35. https://doi.org/10.1007/s10493-015-9914-3
- Brückner A., Wehner K., Neis M., Heethoff M. 2016. Attack and defense in a gamasid-oribatid mite predator-prey experiment - sclerotization outperforms chemical repellency. Acarologia, 56: 451-461. https://doi.org/10.1051/acarologia/20164135
- Bruna T., Hoff K., Stanke M., Lomsadze A., Borodovsky M. 2020. BRAKER2: Automatic Eukaryotic Genome Annotation with GeneMark-EP+ and AUGUSTUS Supported by a Protein Database. bioRxiv. https://doi.org/10.1101/2020.08.10.245134
- Bryon A., Kurlovs A.H., Dermauw W., Greenhalgh R., Riga M., Grbić M., Tirry L., Osakabe M., Vontas J., Clark R.M. 2017. Disruption of a horizontally transferred phytoene desaturase abolishes carotenoid accumulation and diapause in Tetranychus urticae. Proceedings of the National Academy of Sciences, 114: E5871-E5880. https://doi.org/10.1073/pnas.1706865114
- Budelli G., Ni L., Berciu C., van Giesen L., Knecht Z.A., Chang E.C., Kaminski B., Silbering A.F., Samuel A., Klein M. 2019. Ionotropic receptors specify the morphogenesis of phasic sensors controlling rapid thermal preference in Drosophila. Neuron, 101: 738-747. e3. https://doi.org/10.1016/j.neuron.2018.12.022
- Bunnell T., Hanisch K., Hardege J.D., Breithaupt T. 2011. The fecal odor of sick hedgehogs (Erinaceus europaeus) mediates olfactory attraction of the tick Ixodes hexagonus. J Chem Ecol, 37: 340. https://doi.org/10.1007/s10886-011-9936-1
- Cantarel B.L., Coutinho P.M., Rancurel C., Bernard T., Lombard V., Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res, 37: D233-D238. https://doi.org/10.1093/nar/gkn663
- Cao Z., Yu Y., Wu Y., Hao P., Di Z., He Y., Chen Z., Yang W., Shen Z., He X. 2013. The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nature communications, 4: 1-10. https://doi.org/10.1038/ncomms3602
- Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics, 25: 1972-1973. https://doi.org/10.1093/bioinformatics/btp348
- Carlson M., Pagès H. 2019. AnnotationForge: Tools for building SQLite-Based Annotation Data Packages. R package version, 1.
- Chan P.P., Lowe T.M. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. Gene Prediction. Springer. p. 1-14. https://doi.org/10.1007/978-1-4939-9173-0_1
- Charlesworth B. 2012. The effects of deleterious mutations on evolution at linked sites. Genetics, 190: 5-22. https://doi.org/10.1534/genetics.111.134288
- Chikhi R., Medvedev P. 2014. Informed and automated k-mer size selection for genome assembly. Bioinformatics, 30: 31-37. https://doi.org/10.1093/bioinformatics/btt310
- Childers A. Sequenced Arthropod Genomes [Internet]. Manhattan, KS: i5k initiative; [cited].
- Cohen A.C. 1995. Extra-oral digestion in predaceous terrestrial Arthropoda. Annual Rev Entomo, 40: 85-103. https://doi.org/10.1146/annurev.en.40.010195.000505
- Consortium G.O. 2004. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res, 32: D258-D261. https://doi.org/10.1093/nar/gkh036
- Consortium G.O. 2019. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res, 47: D330-D338. https://doi.org/10.1093/nar/gky1055
- Consortium T.G.S. 2008. The genome of the model beetle and pest Tribolium castaneum. Nature, 452: 949. https://doi.org/10.1038/nature06784
- Conway J.R., Lex A., Gehlenborg N. 2017. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics, 33: 2938-2940. https://doi.org/10.1093/bioinformatics/btx364
- Cook C.E., Smith M.L., Telford M.J., Bastianello A., Akam M. 2001. Hox genes and the phylogeny of the arthropods. Curr Biol, 11: 759-763. https://doi.org/10.1016/S0960-9822(01)00222-6
- Cornman R.S., Schatz M.C., Johnston J.S., Chen Y.-P., Pettis J., Hunt G., Bourgeois L., Elsik C., Anderson D., Grozinger C.M. 2010. Genomic survey of the ectoparasitic mite Varroa destructor, a major pest of the honey bee Apis mellifera. BMC Genomics, 11: 602. https://doi.org/10.1186/1471-2164-11-602
- Crescente J.M., Zavallo D., Helguera M., Vanzetti L.S. 2018. MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinf, 19: 348. https://doi.org/10.1186/s12859-018-2376-y
- Crisp A., Boschetti C., Perry M., Tunnacliffe A., Micklem G. 2015. Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol, 16: 1-13. https://doi.org/10.1186/s13059-015-0607-3
- Croset V., Rytz R., Cummins S.F., Budd A., Brawand D., Kaessmann H., Gibson T.J., Benton R. 2010. Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS genetics, 6: e1001064. https://doi.org/10.1371/journal.pgen.1001064
- Dabert M. 2006. DNA markers in the phylogenetics of the Acari. Biological Lett, 43: 97-107.
- Dabert M., Witalinski W., Kazmierski A., Olszanowski Z., Dabert J. 2010. Molecular phylogeny of acariform mites (Acari, Arachnida): Strong conflict between phylogenetic signal and long-branch attraction artifacts. Mol Phylogenet Evol, 56: 222-241. https://doi.org/10.1016/j.ympev.2009.12.020
- Degenhardt J., Köllner T.G., Gershenzon J. 2009. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochem, 70: 1621-1637. https://doi.org/10.1016/j.phytochem.2009.07.030
- Dej K.J., Gerasimova T., Corces V.G., Boeke J.D. 1998. A hotspot for the Drosophila gypsy retroelement in the ovo locus. Nucleic Acids Res, 26: 4019-4024. https://doi.org/10.1093/nar/26.17.4019
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29: 15-21. https://doi.org/10.1093/bioinformatics/bts635
- Domes K., Althammer M., Norton R.A., Scheu S., Maraun M. 2007. The phylogenetic relationship between Astigmata and Oribatida (Acari) as indicated by molecular markers. Exp Appl Acarol, 42: 159-171. https://doi.org/10.1007/s10493-007-9088-8
- Dong X., Armstrong S.D., Xia D., Makepeace B.L., Darby A.C., Kadowaki T. 2017. Draft genome of the honey bee ectoparasitic mite, Tropilaelaps mercedesae, is shaped by the parasitic life history. GigaScience, 6: gix008. https://doi.org/10.1093/gigascience/gix008
- Dong X., Chaisiri K., Xia D., Armstrong S.D., Fang Y., Donnelly M.J., Kadowaki T., McGarry J.W., Darby A.C., Makepeace B.L. 2018. Genomes of trombidid mites reveal novel predicted allergens and laterally transferred genes associated with secondary metabolism. GigaScience, 7: giy127. https://doi.org/10.1093/gigascience/giy127
- dos Santos G., Schroeder A.J., Goodman J.L., Strelets V.B., Crosby M.A., Thurmond J., Emmert D.B., Gelbart W.M., Consortium F. 2015. FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res, 43: D690-D697. https://doi.org/10.1093/nar/gku1099
- Dunlop J., Alberti G. 2008. The affinities of mites and ticks: a review. J Zool Syst Evol Res, 46: 1-18.
- Dunlop J., Selden P. 1998. The early history and phylogeny of the chelicerates. In: Fortey R.A., Thomas R.H., (Eds). Arthropod Relationships. The Systematics Association Special Volume Series. Dordrecht: Springer. p. 221-235. https://doi.org/10.1007/978-94-011-4904-4_17
- Dunlop J.A. 2010. Geological history and phylogeny of Chelicerata. Arthropod Struct Dev, 39: 124-142. https://doi.org/10.1016/j.asd.2010.01.003
- Dunlop J.A., Lamsdell J.C. 2017. Segmentation and tagmosis in Chelicerata. Arthropod Struct Dev, 46: 395-418. https://doi.org/10.1016/j.asd.2016.05.002
- Eddy S.R. 2011. Accelerated profile HMM searches. PLoS Comp Biol, 7: e1002195. https://doi.org/10.1371/journal.pcbi.1002195
- Edgar R.C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32: 1792-1797. https://doi.org/10.1093/nar/gkh340
- Eisenreich W., Bacher A., Arigoni D., Rohdich F. 2004. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cellular and molecular life sciences : CMLS, 61: 1401-1426. https://doi.org/10.1007/s00018-004-3381-z
- Emms D.M., Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol, 16: 157. https://doi.org/10.1186/s13059-015-0721-2
- Eriksson B.J., Fredman D., Steiner G., Schmid A. 2013. Characterisation and localisation of the opsin protein repertoire in the brain and retinas of a spider and an onychophoran. BMC Evol Biol, 13: 186. https://doi.org/10.1186/1471-2148-13-186
- Evans G.O. 1992. Principles of Acarology. Wallingford: CAB International. pp. 563.
- Exner S. 1989. The physiology of the compound eyes of insects and crustaceans. Berlin, Heidelberg: Springer-Verlag GmbH & Co. KG. pp. 177. https://doi.org/10.1007/978-3-642-83595-7
- Faddeeva-Vakhrusheva A., Derks M.F., Anvar S.Y., Agamennone V., Suring W., Smit S., van Straalen N.M., Roelofs D. 2016. Gene family evolution reflects adaptation to soil environmental stressors in the genome of the collembolan Orchesella cincta. Genome Biol Evol, 8: 2106-2117. https://doi.org/10.1093/gbe/evw134
- Faddeeva-Vakhrusheva A., Kraaijeveld K., Derks M.F., Anvar S.Y., Agamennone V., Suring W., Kampfraath A.A., Ellers J., Le Ngoc G., van Gestel C.A. 2017. Coping with living in the soil: the genome of the parthenogenetic springtail Folsomia candida. BMC Genomics, 18: 493. https://doi.org/10.1186/s12864-017-3852-x
- Finnegan D.J. 1989. Eukaryotic transposable elements and genome evolution. Trends Genet, 5: 103-107. https://doi.org/10.1016/0168-9525(89)90039-5
- Flot J.-F., Hespeels B., Li X., Noel B., Arkhipova I., Danchin E.G., Hejnol A., Henrissat B., Koszul R., Aury J.-M. 2013. Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature, 500: 453-457. https://doi.org/10.1038/nature12326
- Fu L., Niu B., Zhu Z., Wu S., Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28: 3150-3152. https://doi.org/10.1093/bioinformatics/bts565
- Gainett G., Ballesteros J.A., Kanzler C.R., Zehms J.T., Zern J.M., Aharon S., Gavish-Regev E., Sharma P.P. 2020. Systemic paralogy and function of retinal determination network homologs in arachnids. BMC Genomics, 21: 811. https://doi.org/10.1186/s12864-020-07149-x
- Gilbert H.J. 2010. The biochemistry and structural biology of plant cell wall deconstruction. Plant Physiol 153: 444-455. https://doi.org/10.1104/pp.110.156646
- Giribet G., Edgecombe G.D. 2019. The phylogeny and evolutionary history of arthropods. Curr Biol, 29: R592-R602. https://doi.org/10.1016/j.cub.2019.04.057
- Gladyshev E.A., Meselson M., Arkhipova I.R. 2008. Massive horizontal gene transfer in bdelloid rotifers. Science, 320: 1210-1213. https://doi.org/10.1126/science.1156407
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q. 2011. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotech, 29: 644. https://doi.org/10.1038/nbt.1883
- Grbić M., Van Leeuwen T., Clark R.M., Rombauts S., Rouzé P., Grbić V., Osborne E.J., Dermauw W., Ngoc P.C.T., Ortego F. 2011. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature, 479: 487-492. https://doi.org/10.1038/nature10640
- Greenhalgh R., Dermauw W., Glas J.J., Rombauts S., Wybouw N., Thomas J., Alba J.M., Pritham E.J., Legarrea S., Feyereisen R. 2020. Genome streamlining in a minute herbivore that manipulates its host plant. Elife, 9: e56689. https://doi.org/10.7554/eLife.56689
- Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol, 59: 307-321. https://doi.org/10.1093/sysbio/syq010
- Gulia-Nuss M., Nuss A.B., Meyer J.M., Sonenshine D.E., Roe R.M., Waterhouse R.M., Sattelle D.B., De La Fuente J., Ribeiro J.M., Megy K. 2016. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nature communications, 7: 1-13.
- Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc., 8: 1494-1512. https://doi.org/10.1038/nprot.2013.084
- Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J., White O., Buell C.R., Wortman J.R. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol., 9: R7. https://doi.org/10.1186/gb-2008-9-1-r7
- Haq M.A. 1993. Symbiotic association of mites and microbes in cellulose degradation. Soil Org Sustain, 4: 81-85.
- Hartmann K., Laumann M., Bergmann P., Heethoff M., Schmelzle S. 2016. Development of the synganglion and morphology of the adult nervous system in the mite Archegozetes longisetosus Aoki (Chelicerata, Actinotrichida, Oribatida). J Morphol, 277: 537-48. https://doi.org/10.1002/jmor.20517
- Harzsch S., Vilpoux K., Blackburn D.C., Platchetzki D., Brown N.L., Melzer R., Kempler K.E., Battelle B.A. 2006. Evolution of arthropod visual systems: development of the eyes and central visual pathways in the horseshoe crab Limulus polyphemus Linnaeus, 1758 (Chelicerata, Xiphosura). Dev Dyn, 235: 2641-2655. https://doi.org/10.1002/dvdy.20866
- Havecker E.R., Gao X., Voytas D.F. 2004. The diversity of LTR retrotransposons. Genome Biol, 5: 1-6. https://doi.org/10.1186/gb-2004-5-6-225
- Heethoff M. 2012. Regeneration of complex oil-gland secretions and its importance for chemical defense in an oribatid mite. J Chem Ecol, 38: 1116-23. https://doi.org/10.1007/s10886-012-0169-8
- Heethoff M., Bergmann P., Laumann M., Norton R.A. 2013. The 20th anniversary of a model mite: A review of current knowledge about Archegozetes longisetosus (Acari, Oribatida). Acarologia, 53: 353-368. https://doi.org/10.1051/acarologia/20132108
- Heethoff M., Bergmann P., Norton R.A. 2006. Karyology and sex determination of oribatid mites. Acarologia, 46: 127-131.
- Heethoff M., Brückner A., Schmelzle S., Schubert M., Bräuer M., Meusinger R., Dötterl S., Norton R.A., Raspotnig G. 2018. Life as a fortress-structure, function, and adaptive values of morphological and chemical defense in the oribatid mite Euphthiracarus reticulatus (Actinotrichida). BMC Zoology, 3: 7. https://doi.org/10.1186/s40850-018-0031-8
- Heethoff M., Koerner L. 2007. Small but powerful: the oribatid mite Archegozetes longisetosus Aoki (Acari, Oribatida) produces disproportionately high forces. J Exp Biol, 210: 3036-3042. https://doi.org/10.1242/jeb.008276
- Heethoff M., Koerner L., Norton R.A., Raspotnig G. 2011a. Tasty but protected-first evidence of chemical defense in oribatid mites. J Chem Ecol, 37: 1037-1043. https://doi.org/10.1007/s10886-011-0009-2
- Heethoff M., Laumann M., Bergmann P. 2007. Adding to the reproductive biology of the parthenogenetic oribatid mite, Archegozetes longisetosus (Acari, Oribatida, Trhypochthoniidae). Turk J Zool, 31: 151-159.
- Heethoff M., Laumann M., Weigmann G., Raspotnig G. 2011b. Integrative taxonomy: Combining morphological, molecular and chemical data for species delineation in the parthenogenetic Trhypochthonius tectorum complex (Acari, Oribatida, Trhypochthoniidae). Front Zool, 8: 2. https://doi.org/10.1186/1742-9994-8-2
- Heethoff M., Norton R.A. 2009. A new use for synchrotron X-ray microtomography: three-dimensional biomechanical modeling of chelicerate mouthparts and calculation of theoretical bite forces. Inver Biol, 128: 332-339. https://doi.org/10.1111/j.1744-7410.2009.00183.x
- Heethoff M., Norton R.A., Raspotnig G. 2016. Once Again: Oribatid Mites and Skin Alkaloids in Poison Frogs. J Chem Ecol, 42: 841-844. https://doi.org/10.1007/s10886-016-0758-z
- Heethoff M., Norton R.A., Scheu S., Maraun M. 2009. Parthenogenesis in Oribatid Mites (Acari, Oribatida): Evolution Without Sex. In: Schön I., Martens K., van Dijk P., (Eds). Lost Sex: The Evolutionary Biology of Parthenogenesis. Dordrecht: Springer. p. 241-257. https://doi.org/10.1007/978-90-481-2770-2_12
- Heethoff M., Rall B.C. 2015. Reducible defence: chemical protection alters the dynamics of predator-prey interactions. Chemoecology, 25: 53-61. https://doi.org/10.1007/s00049-014-0184-z
- Heethoff M., Raspotnig G. 2012. Expanding the ′enemy-free space′ for oribatid mites: evidence for chemical defense of juvenile Archegozetes longisetosus against the rove beetle Stenus juno. Exp Appl Acarol, 56: 93-97. https://doi.org/10.1007/s10493-011-9501-1
- Heidemann K., Scheu S., Ruess L., Maraun M. 2011. Molecular detection of nematode predation and scavenging in oribatid mites: Laboratory and field experiments. Soil Biol Biochem, 43: 229-236. https://doi.org/10.1016/j.soilbio.2011.07.015
- Heingård M., Turetzek N., Prpic N.-M., Janssen R. 2019. FoxB, a new and highly conserved key factor in arthropod dorsal-ventral (DV) limb patterning. EvoDevo, 10: 1-16. https://doi.org/10.1186/s13227-019-0141-6
- Hoffmann A., Thimm T., Dröge M., Moore E.R., Munch J.C., Tebbe C.C. 1998. Intergeneric transfer of conjugative and mobilizable plasmids harbored by Escherichia coli in the gut of the soil microarthropod Folsomia candida (Collembola). Appl Environ Microbiol, 64: 2652-2659. https://doi.org/10.1128/AEM.64.7.2652-2659.1998
- Holland P., Hogan B. 1988. Expression of homeo box genes during mouse development: a review. Gene Devol, 2: 773-782. https://doi.org/10.1101/gad.2.7.773
- Hoy M.A., Waterhouse R.M., Wu K., Estep A.S., Ioannidis P., Palmer W.J., Pomerantz A.F., Simao F.A., Thomas J., Jiggins F.M. 2016. Genome sequencing of the phytoseiid predatory mite Metaseiulus occidentalis reveals completely atomized Hox genes and superdynamic intron evolution. Genome Biol Evol, 8: 1762-1775. https://doi.org/10.1093/gbe/evw048
- Hrycaj S.M., Wellik D.M. 2016. Hox genes and evolution. F1000Research, 5. https://doi.org/10.12688/f1000research.7663.1
- Huerta-Cepas J., Forslund K., Coelho L.P., Szklarczyk D., Jensen L.J., Von Mering C., Bork P. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol., 34: 2115-2122. https://doi.org/10.1093/molbev/msx148
- Hughes C.L., Kaufman T.C. 2002. Hox genes and the evolution of the arthropod body plan. Evol Dev, 4: 459-499. https://doi.org/10.1046/j.1525-142X.2002.02034.x
- Jeyaprakash A., Hoy M.A. 2009. First divergence time estimate of spiders, scorpions, mites and ticks (subphylum: Chelicerata) inferred from mitochondrial phylogeny. Exp Appl Acarol, 47: 1-18. https://doi.org/10.1007/s10493-008-9203-5
- Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T. 2007. KEGG for linking genomes to life and the environment. Nucleic Acids Res, 36: D480-D484. https://doi.org/10.1093/nar/gkm882
- Kanehisa M., Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res, 28: 27-30. https://doi.org/10.1093/nar/28.1.27
- Kanehisa M., Sato Y., Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol, 428: 726-731. https://doi.org/10.1016/j.jmb.2015.11.006
- Katoh K., Standley D.M. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol, 30: 772-780. https://doi.org/10.1093/molbev/mst010
- Keilwagen J., Hartung F., Grau J. 2019. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene Prediction. Amsterdam: Springer. p. 161-177. https://doi.org/10.1007/978-1-4939-9173-0_9
- Kitchen S.A., Crowder C.M., Poole A.Z., Weis V.M., Meyer E. 2015. De novo assembly and characterization of four anthozoan (Cnidaria) transcriptomes. G3, 5: 2441-2452. https://doi.org/10.1534/g3.115.020164
- Klimov P.B., OConnor B. 2013. Is permanent parasitism reversible?-Critical evidence from early evolution of house dust mites. Syst Biol, 62: 411-423. https://doi.org/10.1093/sysbio/syt008
- Klimov P.B., OConnor B.M., Chetverikov P.E., Bolton S.J., Pepato A.R., Mortazavi A.L., Tolstikov A.V., Bauchan G.R., Ochoa R. 2018. Comprehensive phylogeny of acariform mites (Acariformes) provides insights on the origin of the four-legged mites (Eriophyoidea), a long branch. Mol Phylogenet Evol, 119: 105-117. https://doi.org/10.1016/j.ympev.2017.10.017
- Knecht Z.A., Silbering A.F., Ni L., Klein M., Budelli G., Bell R., Abuin L., Ferrer A.J., Samuel A.D., Benton R. 2016. Distinct combinations of variant ionotropic glutamate receptors mediate thermosensation and hygrosensation in Drosophila. eLife, 5: e17879. https://doi.org/10.7554/eLife.17879
- Kocot K.M., Citarella M.R., Moroz L.L., Halanych K.M. 2013. PhyloTreePruner: a phylogenetic tree-based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinform. Online, 9: 429-435. https://doi.org/10.4137/EBO.S12813
- Kohany O., Gentles A.J., Hankus L., Jurka J. 2006. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics, 7: 1-7. https://doi.org/10.1186/1471-2105-7-474
- Koller L.M., Wirth S., Raspotnig G. 2012. Geranial-rich oil gland secretions: a common phenomenon in the Histiostomatidae (Acari, Astigmata)? Int J Acarol, 38: 420-426. https://doi.org/10.1080/01647954.2012.662247
- Königsmann T., Turetzek N., Pechmann M., Prpic N.-M. 2017. Expression and function of the zinc finger transcription factor Sp6-9 in the spider Parasteatoda tepidariorum. Dev Genes Evol, 227: 389-400. https://doi.org/10.1007/s00427-017-0595-2
- Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res, 27: 722-736. https://doi.org/10.1101/gr.215087.116
- Koyanagi M., Nagata T., Katoh K., Yamashita S., Tokunaga F. 2008. Molecular evolution of arthropod color vision deduced from multiple opsin genes of jumping spiders. J Mol Evol, 66: 130-137. https://doi.org/10.1007/s00239-008-9065-9
- Kück P., Meusemann K. 2010. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol., 56: 1115-1118. https://doi.org/10.1016/j.ympev.2010.04.024
- Kuwahara Y. 2004. Chemical ecology of astigmatid mites. In: Cardé R.T., Millar J.G., (Eds). Advances in Insect Chemical Ecology. Cambridge: Cambridge University Press. p. 76-109. https://doi.org/10.1017/CBO9780511542664.004
- Kuwahara Y., Ibi T., Nakatani Y., Ryouno A., Mori N., Sakata T., Okabe K., Tagami K., Kurosa K. 2001. Chemical ecology of astigmatid mites LIX. Neral, the alarm pheromone of Schwiebea elongata (Banks)(Acari: Acaridae). J Acarol Soc Japan, 10: 19-25. https://doi.org/10.2300/acari.10.19
- Laetsch D.R., Blaxter M.L. 2017. BlobTools: Interrogation of genome assemblies. F1000Research, 6: 1287. https://doi.org/10.12688/f1000research.12232.1
- Lanfear R., Frandsen P.B., Wright A.M., Senfeld T., Calcott B. 2016. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol., 34: 772-773. https://doi.org/10.1093/molbev/msw260
- Latgé J.P. 2007. The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol, 66: 279-290. https://doi.org/10.1111/j.1365-2958.2007.05872.x
- Lawrence J.G. 1997. Selfish operons and speciation by gene transfer. Trends Microbiol, 5: 355-359. https://doi.org/10.1016/S0966-842X(97)01110-4
- Lefort V., Longueville J.-E., Gascuel O. 2017. SMS: smart model selection in PhyML. Mol Biol Evol, 34: 2422-2424. https://doi.org/10.1093/molbev/msx149
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34: 3094-3100. https://doi.org/10.1093/bioinformatics/bty191
- Li H., Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25: 1754-1760. https://doi.org/10.1093/bioinformatics/btp324
- Li W.-N., Xue X.-F. 2019. Mitochondrial genome reorganization provides insights into the relationship between oribatid mites and astigmatid mites (Acari: Sarcoptiformes: Oribatida). Zoolo J Lin Soc, 187: 585-598. https://doi.org/10.1093/zoolinnean/zlz044
- Liana M., Witaliński W. 2005. Sperm structure and phylogeny of Astigmata. J Morphol, 265: 318-324. https://doi.org/10.1002/jmor.10361
- Liu W., Zhang R., Tian N., Xu X., Cao Y., Xian M., Liu H. 2015. Utilization of alkaline phosphatase PhoA in the bioproduction of geraniol by metabolically engineered Escherichia coli. Bioengineered, 6: 288-293. https://doi.org/10.1080/21655979.2015.1062188
- Lozano-Fernandez J., Tanner A.R., Giacomelli M., Carton R., Vinther J., Edgecombe G.D., Pisani D. 2019. Increasing species sampling in chelicerate genomic-scale datasets provides support for monophyly of Acari and Arachnida. Nature Comm, 10: 1-8. https://doi.org/10.1038/s41467-019-10244-7
- Luxton M. 1972. Studies on oribatid mites of a Danish beech wood soil .1. Nutritional Biology. Pedobiologia, 12: 434-463.
- Luxton M. 1979. Food and energy processing by oribatid mites. Rev Ecol Biol Sol, 16: 103-111.
- Luxton M. 1981. Studies on the oribatid mites of a Danish beech wood soil .7. Energy Budgets. Pedobiologia, 22: 77-111.
- Luxton M. 1982. The bology of mites from beech woodland soil. Pedobiologia, 23: 1-8.
- Madge D. 1965. Further studies on the behaviour of Belba geniculosa Oudms. in relation to various environmental stimuli. Acarologia, 7: 744-757.
- Maraun M., Erdmann G., Fischer B.M., Pollierer M.M., Norton R.A., Schneider K., Scheu S. 2011. Stable isotopes revisited: Their use and limits for oribatid mite trophic ecology. Soil Biol Biochem, 43: 877-882. https://doi.org/10.1016/j.soilbio.2011.01.003
- Maraun M., Heethoff M., Schneider K., Scheu S., Weigmann G., Cianciolo J., Thomas R.H., Norton R.A. 2004. Molecular phylogeny of oribatid mites (Oribatida, Acari): evidence for multiple radiations of parthenogenetic lineages. Exp Appl Acarol, 33: 183-201. https://doi.org/10.1023/B:APPA.0000032956.60108.6d
- Maraun M., Schatz H., Scheu S. 2007. Awesome or ordinary? Global diversity patterns of oribatid mites. Ecography, 30: 209-216. https://doi.org/10.1111/j.0906-7590.2007.04994.x
- Maraun M., Scheu S. 2000. The structure of oribatid mite communities (Acari, Oribatida): Patterns, mechanisms and implications for future research. Ecography, 23: 374-383. https://doi.org/10.1034/j.1600-0587.2000.d01-1647.x
- Marçais G., Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics, 27: 764-770. https://doi.org/10.1093/bioinformatics/btr011
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J, 17: 10-12. https://doi.org/10.14806/ej.17.1.200
- Mayer W.E., Schuster L.N., Bartelmes G., Dieterich C., Sommer R.J. 2011. Horizontal gene transfer of microbial cellulases into nematode genomes is associated with functional assimilation and gene turnover. BMC Evol Biol, 11: 1-10. https://doi.org/10.1186/1471-2148-11-13
- McKenna D.D., Scully E.D., Pauchet Y., Hoover K., Kirsch R., Geib S.M., Mitchell R.F., Waterhouse R.M., Ahn S.-J., Arsala D. 2016. Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle-plant interface. Genome Biol, 17: 1-18. https://doi.org/10.1186/s13059-016-1088-8
- McKenna D.D., Shin S., Ahrens D., Balke M., Beza-Beza C., Clarke D.J., Donath A., Escalona H.E., Friedrich F., Letsch H. 2019. The evolution and genomic basis of beetle diversity. Proceedings of the National Academy of Sciences, 116: 24729-24737. https://doi.org/10.1073/pnas.1909655116
- Mitreva M., Smant G., Helder J. 2009. Role of horizontal gene transfer in the evolution of plant parasitism among nematodes. Horizontal Gene Transfer. Amsterdam: Springer. p. 517-535. https://doi.org/10.1007/978-1-60327-853-9_30
- Miziorko H.M. 2011. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys, 505: 131-143. https://doi.org/10.1016/j.abb.2010.09.028
- Montell C. 2009. A taste of the Drosophila gustatory receptors. Curr Opin Neurobiol, 19: 345-353. https://doi.org/10.1016/j.conb.2009.07.001
- Morita A., Mori N., Nishida R., Hirai N., Kuwahara Y. 2004. Neral biosynthesis via the mevalonate pathway, evidenced by D-glucose-1-13C feeding in Carpoglyphus lactis and 13C incorporation into other opisthonotal gland exudates. J Pest Sci, 29: 27-32. https://doi.org/10.1584/jpestics.29.27
- Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res, 35: W182-W185. https://doi.org/10.1093/nar/gkm321
- Muller H.J. 1964. The relation of recombination to mutaional advance. Mutation Res 106: 2-9. https://doi.org/10.1016/0027-5107(64)90047-8
- Nagata T., Koyanagi M., Tsukamoto H., Terakita A. 2010. Identification and characterization of a protostome homologue of peropsin from a jumping spider. J Comp Physiol A, 196: 51. https://doi.org/10.1007/s00359-009-0493-9
- Nagy L.G., Merényi Z., Hegedüs B., Bálint B. 2020. Novel phylogenetic methods are needed for understanding gene function in the era of mega-scale genome sequencing. Nucleic Acids Res, 48: 2209-2219. https://doi.org/10.1093/nar/gkz1241
- Ngoc P.C.T., Greenhalgh R., Dermauw W., Rombauts S., Bajda S., Zhurov V., Grbić M., Van de Peer Y., Van Leeuwen T., Rouze P. 2016. Complex evolutionary dynamics of massively expanded chemosensory receptor families in an extreme generalist chelicerate herbivore. Genome Biol Evol, 8: 3323-3339. https://doi.org/10.1093/gbe/evw249
- Nguyen L.-T., Schmidt H.A., Von Haeseler A., Minh B.Q. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol, 32: 268-274. https://doi.org/10.1093/molbev/msu300
- Niimura Y., Nei M. 2005. Evolutionary dynamics of olfactory receptor genes in fishes and tetrapods. PNAS, 102: 6039-6044. https://doi.org/10.1073/pnas.0501922102
- Noge K., Kato M., Iguchi T., Mori N., Nishida R., Kuwahara Y. 2005. Biosynthesis of neral in Carpoglyphus lactis (Acari: Carpoglyphidae) and detection of its key enzyme, geraniol dehydrogenase, by electrophoresis. J Acarol Soc Japan, 14: 75-81. https://doi.org/10.2300/acari.14.75
- Noge K., Kato M., Mori N., Kataoka M., Tanaka C., Yamasue Y., Nishida R., Kuwahara Y. 2008. Geraniol dehydrogenase, the key enzyme in biosynthesis of the alarm pheromone, from the astigmatid mite Carpoglyphus lactis (Acari: Carpoglyphidae). The FEBS journal, 275: 2807-2817. https://doi.org/10.1111/j.1742-4658.2008.06421.x
- Norton R.A. 1994. Evolutionary aspects of oribatid mite life histories and consequences for the origin of the Astigmata. In: Houck M.A., (Ed). Mites: Ecological and Evolutionary Analyses of Life-History Patterns. New York: Chapman & Hall. p. 99-135. https://doi.org/10.1007/978-1-4615-2389-5_5
- Norton R.A. 1998. Morphological evidence for the evolutionary origin of Astigmata (Acari : Acariformes). Exp Appl Acarol, 22: 559-594.
- Norton R.A. Year of Conferenc. Holistic acarology and ultimate causes: examples from the oribatid mites. In: Morales-Malacara J.B., Behan-Pelletier V., Ueckermann E., Perez T.M., Estrada-Venegas E.G., Badii M., (Eds). Acarology XI: Proceedings of the International Congress.; Mexico: Sociedad Latinoamericana de Acarologia. p. 3-20.
- Norton R.A., Franklin E. 2018. Paraquanothrus n. gen. from freshwater rock pools in the USA, with new diagnoses of Aquanothrus, Aquanothrinae, and Ameronothridae (Acari, Oribatida). Acarologia, 58: 557-627. https://doi.org/10.24349/acarologia/20184258
- Norton R.A., Fuangarworn M. 2015. Nanohystricidae n. fam., an unusual, plesiomorphic enarthronote mite family endemic to New Zealand (Acari, Oribatida). Zootaxa, 4027: 151-204. https://doi.org/10.11646/zootaxa.4027.2.1
- Norton R.A., Kethley J.B., Johnston D.E., O'Connor B.M. 1993. Phylogenetic perspectives on genetic systems and reproductive modes of mites. In: Wrensch D., Ebbert M., (Eds). Evolution and Diversity of Sex Ratio in Insects and Mites. London: Chapman & Hall. p. 8-99. https://doi.org/10.1007/978-1-4684-1402-8_2
- Norton R.A., Palmer S.C. 1991. The distribution, mechanisms and evolutionary significance of parthenogenesis in oribatid mites. In: Schuster R., Murphy P.W., (Eds). The Acari - Reproduction, Development and Life-History Strategies. London: Chapman & Hall. p. 107-136. https://doi.org/10.1007/978-94-011-3102-5_7
- Nuzhdin S.V., Petrov D.A. 2003. Transposable elements in clonal lineages: lethal hangover from sex. Biol J Lin Soc, 79: 33-41. https://doi.org/10.1046/j.1095-8312.2003.00188.x
- Oldfield E., Lin F.Y. 2012. Terpene biosynthesis: modularity rules. Angew Chem Int Ed Engl, 51: 1124-1137. https://doi.org/10.1002/anie.201103110
- Oliver Jr J.H. 1983. Chromosomes, genetic variance and reproductive strategies among mites and ticks. Bull Entomol Soc Am, 29: 8-17. https://doi.org/10.1093/besa/29.2.8
- Oswald M., Fischer M., Dirninger N., Karst F. 2007. Monoterpenoid biosynthesis in Saccharomyces cerevisiae. FEMS Yeast Res 7: 413-421. https://doi.org/10.1111/j.1567-1364.2006.00172.x
- Oxley P.R., Ji L., Fetter-Pruneda I., McKenzie S.K., Li C., Hu H., Zhang G., Kronauer D.J. 2014. The genome of the clonal raider ant Cerapachys biroi. Curr Biol, 24: 451-458. https://doi.org/10.1016/j.cub.2014.01.018
- Pace R.M., Grbić M., Nagy L.M. 2016. Composition and genomic organization of arthropod Hox clusters. EvoDevo, 7: 11. https://doi.org/10.1186/s13227-016-0048-4
- Pachl P., Domes K., Schulz G., Norton R.A., Scheu S., Schaefer I., Maraun M. 2012. Convergent evolution of defense mechanisms in oribatid mites (Acari, Oribatida) shows no "ghosts of predation past″. Mol Phylogenet Evol, 65: 412-420. https://doi.org/10.1016/j.ympev.2012.06.030
- Palmer J., Stajich J. 2017. Funannotate: eukaryotic genome annotation pipeline. https://funannotate.readthedocs.io/en/latest/ .
- Palmer M., Bantle J., Guo X., Fargoxy1 W.S. 1994. Genome size and organization in the ixodid tick Amblyomma americanum (L.). Insect Mol Biol, 3: 57-62. https://doi.org/10.1111/j.1365-2583.1994.tb00151.x
- Palmer S.C., Norton R.A. 1992. Genetic diversity in thelytokous oribatid mites (Acari; Acariformes: Desmonomata). Biochem Syst Ecol, 20: 219-231. https://doi.org/10.1016/0305-1978(92)90056-J
- Panfilio K.A., Jentzsch I.M.V., Benoit J.B., Erezyilmaz D., Suzuki Y., Colella S., Robertson H.M., Poelchau M.F., Waterhouse R.M., Ioannidis P. 2019. Molecular evolutionary trends and feeding ecology diversification in the Hemiptera, anchored by the milkweed bug genome. Genome Biol, 20: 64. https://doi.org/10.1186/s13059-019-1660-0
- Patten W. 1887. Eyes of molluscs and arthropods. J Morphol, 1: 67-92. https://doi.org/10.1002/jmor.1050010105
- Pepato A., Klimov P. 2015. Origin and higher-level diversification of acariform mites-evidence from nuclear ribosomal genes, extensive taxon sampling, and secondary structure alignment. BMC Evol Biol, 15: 178. https://doi.org/10.1186/s12862-015-0458-2
- Petersen M., Armisén D., Gibbs R.A., Hering L., Khila A., Mayer G., Richards S., Niehuis O., Misof B. 2019. Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Evol. Biol., 19: 11. https://doi.org/10.1186/s12862-018-1324-9
- Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L. 2017. Differential analysis of RNA-seq incorporating quantification uncertainty. Nature Meth 14: 687. https://doi.org/10.1038/nmeth.4324
- Price M.N., Dehal P.S., Arkin A.P. 2010. FastTree 2 - approximately Maximum-Likelihood trees for large alignments. PLoS One, 5: e9490. https://doi.org/10.1371/journal.pone.0009490
- Pruitt K.D., Tatusova T., Maglott D.R. 2005. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res, 33: D501-D504. https://doi.org/10.1093/nar/gki025
- Quinlan A.R., Hall I.M. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26: 841-842. https://doi.org/10.1093/bioinformatics/btq033
- Quinodoz S.A., Bhat P., Chovanec P., Jachowicz J.W., Ollikainen N., Detmar E., Soehalim E., Guttman M. 2022. SPRITE: a genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding. Nat Protoc: 1-41. https://doi.org/10.1038/s41596-021-00633-y
- R_Core_Team. 2019. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2019.
- Ramírez F., Bhardwaj V., Arrigoni L., Lam K.C., Grüning B.A., Villaveces J., Habermann B., Akhtar A., Manke T. 2018. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nature communications, 9: 1-15. https://doi.org/10.1038/s41467-017-02525-w
- Raspotnig G. 2006. Chemical alarm and defence in the oribatid mite Collohmannia gigantea (Acari: Oribatida). Exp Appl Acarol, 39: 177-194. https://doi.org/10.1007/s10493-006-9015-4
- Raspotnig G. Year of Conferenc. Oil gland secretions in Oribatida (Acari). In: Sabelis M.W., Bruin J., (Eds). Trends in Acarology; Dordrecht: Springer. p. 235-239. https://doi.org/10.1007/978-90-481-9837-5_38
- Raspotnig G., Kaiser R., Stabentheiner E., Leis H.J. 2008. Chrysomelidial in the Opisthonotal Glands of the Oribatid Mite, Oribotritia berlesei. J Chem Ecol, 34: 1081-1088. https://doi.org/10.1007/s10886-008-9508-1
- Raspotnig G., Norton R.A., Heethoff M. 2011. Oribatid mites and skin alkaloids in poison frogs. Biol Lett, 7: 555-556. https://doi.org/10.1098/rsbl.2010.1113
- Raspotnig G., Schuster R., Krisper G. 2004. Citral in oil gland secretions of Oribatida (Acari): a key component for phylogenetic analyses. Abh Ber Naturkundemus Görlitz, 76: 43-50.
- Rawlings N.D., Barrett A.J., Bateman A. 2010. MEROPS: the peptidase database. Nucleic Acids Res, 38: D227-D233. https://doi.org/10.1093/nar/gkp971
- Regier J.C., Shultz J.W., Zwick A., Hussey A., Ball B., Wetzer R., Martin J.W., Cunningham C.W. 2010. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature, 463: 1079-1083. https://doi.org/10.1038/nature08742
- Renschler G., Richard G., Valsecchi C.I.K., Toscano S., Arrigoni L., Ramírez F., Akhtar A. 2019. Hi-C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling. Genes & development, 33: 1591-1612. https://doi.org/10.1101/gad.328971.119
- Richards S. 2019. Arthropod genome sequencing and assembly strategies. Insect Genomics: 1-14. https://doi.org/10.1007/978-1-4939-8775-7_1
- Rider S.D., Morgan M.S., Arlian L.G. 2015. Draft genome of the scabies mite. Parasite Vectors, 8: 1-14. https://doi.org/10.1186/s13071-015-1198-2
- Riha G. 1951. Zur Ökologie der Oribatiden in Kalksteinböden. Zoolo Jahrb, 80: 407-450.
- Roach M.J., Schmidt S.A., Borneman A.R. 2018. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC bioinformatics, 19: 1-10. https://doi.org/10.1186/s12859-018-2485-7
- Robertson H.M., Wanner K.W. 2006. The chemoreceptor superfamily in the honey bee, Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res, 16: 1395-1403. https://doi.org/10.1101/gr.5057506
- Robertson H.M., Warr C.G., Carlson J.R. 2003. Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. PNAS, 100: 14537-14542. https://doi.org/10.1073/pnas.2335847100
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. 2011. Integrative genomics viewer. Nat. Biotechnol., 29: 24-26. https://doi.org/10.1038/nbt.1754
- Rognes T., Flouri T., Nichols B., Quince C., Mahé F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ, 4: e2584. https://doi.org/10.7717/peerj.2584
- Rytz R., Croset V., Benton R. 2013. Ionotropic receptors (IRs): chemosensory ionotropic glutamate receptors in Drosophila and beyond. Insect Biochem Mol Biol, 43: 888-897. https://doi.org/10.1016/j.ibmb.2013.02.007
- Sakata T. 1997. Natural Chemistry of Mite Secretions. [Kyoto, Japan]: Kyoto University. pp. 153.
- Sakata T., Norton R.A. 2001. Opisthonotal gland chemistry of early-derivative oribatid mites (Acari) and its relevance to systematic relationships of Astigmata. Int J Acarol, 27: 281-292. https://doi.org/10.1080/01647950108684268
- Sakata T., Norton R.A. 2003. Opisthonotal gland chemistry of a middle-derivative oribatid mite, Archegozetes longisetosus (Acari : Trhypochthoniidae). Int J Acarol, 29: 345-350. https://doi.org/10.1080/01647950308684351
- Sakata T., Tagami K., Kuwahara Y. 1995. Chemical ecology of oribatid mites. I. Oil gland components of Hydronothrus crispus Aoki. J Acarol Soc Japan, 4: 69-75. https://doi.org/10.2300/acari.4.69
- Samadi L., Schmid A., Eriksson B.J. 2015. Differential expression of retinal determination genes in the principal and secondary eyes of Cupiennius salei Keyserling (1877). EvoDevo, 6: 16. https://doi.org/10.1186/s13227-015-0010-x
- Sánchez-Gracia A., Vieira F., Rozas J. 2009. Molecular evolution of the major chemosensory gene families in insects. Heredity, 103: 208-216. https://doi.org/10.1038/hdy.2009.55
- Sánchez-Gracia A., Vieira F.G., Almeida F.C., Rozas J. 2011. Comparative genomics of the major chemosensory gene families in Arthropods. eLS. https://doi.org/10.1002/9780470015902.a0022848
- Santos V.T., Ribeiro L., Fraga A., de Barros C.M., Campos E., Moraes J., Fontenele M.R., Araujo H.M., Feitosa N.M., Logullo C. 2013. The embryogenesis of the tick Rhipicephalus (Boophilus) microplus: the establishment of a new chelicerate model system. Genesis, 51: 803-818. https://doi.org/10.1002/dvg.22717
- Saporito R.A., Donnelly M.A., Norton R.A., Garraffo H.M., Spande T.F., Daly J.W. 2007. Oribatid mites as a major dietary source for alkaloids in poison frogs. PNAS, 104: 8885-8890. https://doi.org/10.1073/pnas.0702851104
- Saporito R.A., Spande T.F., Garraffo H.M., Donnelly M.A. 2009. Arthropod alkaloids in poison frogs: A review of the dietary hypothesis. Heterocycles, 79: 277-297. https://doi.org/10.3987/REV-08-SR(D)11
- Schaefer I., Norton R.A., Scheu S., Maraun M. 2010. Arthropod colonization of land - Linking molecules and fossils in oribatid mites (Acari, Oribatida). Mol Phylogenet Evol, 57: 113-121. https://doi.org/10.1016/j.ympev.2010.04.015
- Schmelzle S., Blüthgen N. 2019. Under pressure: force resistance measurements in box mites (Actinotrichida, Oribatida). Front Zool, 16: 24. https://doi.org/10.1186/s12983-019-0325-x
- Schneider K., Maraun M. 2005. Feeding preferences among dark pigmented fungal taxa ("Dematiacea") indicate limited trophic niche differentiation of oribatid mites (Oribatida, Acari). Pedobiologia, 49: 61-67. https://doi.org/10.1016/j.pedobi.2004.07.010
- Schneider K., Migge S., Norton R.A., Scheu S., Langel R., Reineking A., Maraun M. 2004a. Trophic niche differentiation in soil microarthropods (Oribatida, Acari): evidence from stable isotope ratios (N-15/N-14). Soil Biol Biochem, 36: 1769-1774. https://doi.org/10.1016/j.soilbio.2004.04.033
- Schneider K., Renker C., Scheu S., Maraun M. 2004b. Feeding biology of oribatid mites: a minireview. Phytophaga, 14: 247-256.
- Schomburg C., Turetzek N., Schacht M.I., Schneider J., Kirfel P., Prpic N.-M., Posnien N. 2015. Molecular characterization and embryonic origin of the eyes in the common house spider Parasteatoda tepidariorum. EvoDevo, 6: 1-14. https://doi.org/10.1186/s13227-015-0011-9
- Schön I., Martens K., van Dijk P. 2009. Lost Sex - The Evolutionary Biology of Parthenogenesis. Dordrecht: Springer. https://doi.org/10.1007/978-90-481-2770-2
- Schwager E.E., Schönauer A., Leite D.J., Sharma P.P., McGregor A.P. 2015. Chelicerata. Amsterdam: Springer. https://doi.org/10.1007/978-3-7091-1865-8_5
- Schwager E.E., Sharma P.P., Clarke T., Leite D.J., Wierschin T., Pechmann M., Akiyama-Oda Y., Esposito L., Bechsgaard J., Bilde T. 2017. The house spider genome reveals an ancient whole-genome duplication during arachnid evolution. BMC Biology, 15: 1-27. https://doi.org/10.1186/s12915-017-0399-x
- Senthilan P.R., Grebler R., Reinhard N., Rieger D., Helfrich-Förster C. 2019. Role of rhodopsins as circadian photoreceptors in the Drosophila melanogaster. Biology, 8: 6. https://doi.org/10.3390/biology8010006
- Senthilan P.R., Helfrich-Förster C. 2016. Rhodopsin 7-the unusual rhodopsin in Drosophila. PeerJ, 4: e2427. https://doi.org/10.7717/peerj.2427
- Sharma P.P., Schwager E.E., Extavour C.G., Giribet G. 2012. Hox gene expression in the harvestman Phalangium opilio reveals divergent patterning of the chelicerate opisthosoma. Evol Dev, 14: 450-463. https://doi.org/10.1111/j.1525-142X.2012.00565.x
- Sharma P.P., Tarazona O.A., Lopez D.H., Schwager E.E., Cohn M.J., Wheeler W.C., Extavour C.G. 2015. A conserved genetic mechanism specifies deutocerebral appendage identity in insects and arachnids. Proc R Soc Lond Biol, 282: 20150698. https://doi.org/10.1098/rspb.2015.0698
- Shen W.L., Kwon Y., Adegbola A.A., Luo J., Chess A., Montell C. 2011. Function of rhodopsin in temperature discrimination in Drosophila. Science, 331: 1333-1336. https://doi.org/10.1126/science.1198904
- Shimano S., Sakata T., Mizutani Y., Kuwahara Y., Aoki J.-i. 2002. Geranial: the alarm pheromone in the nymphal stage of the oribatid mite, Nothrus palustris. J Chem Ecol, 28: 1831-1837. https://doi.org/10.1023/A:1020517319363
- Shimizu N., Sakata D., Schmelz E.A., Mori N., Kuwahara Y. 2017. Biosynthetic pathway of aliphatic formates via a Baeyer-Villiger oxidation in mechanism present in astigmatid mites. PNAS, 114: 2616-2621. https://doi.org/10.1073/pnas.1612611114
- Shingate P., Ravi V., Prasad A., Tay B.-H., Garg K.M., Chattopadhyay B., Yap L.-M., Rheindt F.E., Venkatesh B. 2020. Chromosome-level assembly of the horseshoe crab genome provides insights into its genome evolution. Nature Comm, 11: 1-13. https://doi.org/10.1038/s41467-020-16180-1
- Shultz J.W. 2007. A phylogenetic analysis of the arachnid orders based on morphological characters. Zoolo J Lin Soc, 150: 221-265. https://doi.org/10.1111/j.1096-3642.2007.00284.x
- Shumate A., Salzberg S.L. 2021. Liftoff: accurate mapping of gene annotations. Bioinformatics, 37: 1639-1643. https://doi.org/10.1093/bioinformatics/btaa1016
- Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034-1050. https://doi.org/10.1101/gr.3715005
- Siepel H., de Ruiter-Dijkman E.M. 1993. Feeding guilds of oribatid mites based on their carbohydrase activities. Soil Biol Biochem, 25: 1491-1497. https://doi.org/10.1016/0038-0717(93)90004-U
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31: 3210-3212. https://doi.org/10.1093/bioinformatics/btv351
- Smit A., Hubley R., Green P. RepeatMasker Open-3.0 [Internet]. [cited]. Available from: http://www.repeatmasker.org .
- Smit A.F., Hubley R. 2008. RepeatModeler Open-1.0.
- Smrž J. 1992. Some adaptive features in the microanatomy of moss-dwelling oribatid mites (Acari: Oribatida) with respect to their ontogenetical development. Pedobiologia, 36: 306-320.
- Smrž J. 2000. A modified test for chitinase and cellulase activity in soil mites. Pedobiologia, 44: 186-189. https://doi.org/10.1078/S0031-4056(04)70037-2
- Smrž J., Čatská V. 2010. Mycophagous mites and their internal associated bacteria cooperate to digest chitin in soil. Symbiosis, 52: 33-40. https://doi.org/10.1007/s13199-010-0099-6
- Smrž J., Norton R.A. 2004. Food selection and internal processing in Archegozetes longisetosus (Acari : Oribatida). Pedobiologia, 48: 111-120. https://doi.org/10.1016/j.pedobi.2003.09.003
- Stefaniak O. 1976. The microflora of the alimentary canal of Achipteria coleoptrata (Acarina, Oribatei). Pedobiologia, 16.
- Stefaniak O. 1981. The effect of fungal diet on the development of Oppia nitens (Acari, Oribatei) and on the microflora of its alimentary tract. Pedobiologia, 21: 202-210.
- Sun H., Ding J., Piednoël M., Schneeberger K. 2018. findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics, 34: 550-557. https://doi.org/10.1093/bioinformatics/btx637
- Suzuki S., Kakuta M., Ishida T., Akiyama Y. 2014. GHOSTX: an improved sequence homology search algorithm using a query suffix array and a database suffix array. PloS One, 9: e103833. https://doi.org/10.1371/journal.pone.0103833
- Telford M.J., Thomas R.H. 1998. Expression of homeobox genes shows chelicerate arthropods retain their deutocerebral segment. PNAS, 95: 10671-10675. https://doi.org/10.1073/pnas.95.18.10671
- Ter-Hovhannisyan V., Lomsadze A., Chernoff Y.O., Borodovsky M. 2008. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res., 18: 1979-1990. https://doi.org/10.1101/gr.081612.108
- Thiel T., Brechtel A., Brückner A., Heethoff M., Drossel B. 2018. The effect of reservoir-based chemical defense on predator-prey dynamics. Theo Ecol, 12: 365-378. https://doi.org/10.1007/s12080-018-0402-3
- Thomas G.W., Dohmen E., Hughes D.S., Murali S.C., Poelchau M., Glastad K., Anstead C.A., Ayoub N.A., Batterham P., Bellair M. 2020. Gene content evolution in the arthropods. Genome Biol 21: 1-14. https://doi.org/10.1186/s13059-019-1925-7
- Thomas R.H. Year of Conferenc. Mites as models in development and genetics. In: Bernini F., Nannelli R., Nuzzaci G., de Lillo E., (Eds). Acarid Phylogeny and Evolution: Adaptation in Mites and Ticks: Proceedings of the IV Symposium of the European Association of Acarologists; Dordrecht: Kluwer Academic Publishers. p. 21-26.
- Thorpe P., Escudero-Martinez C.M., Cock P.J., Eves-van den Akker S., Bos J.I. 2018. Shared transcriptional control and disparate gain and loss of aphid parasitism genes. Genome Biol. Evol., 10: 2716-2733. https://doi.org/10.1093/gbe/evy183
- Trägårdh I. 1933. Methods of automatic collecting for studying the fauna of the soil. Bull Entomol Res, 24: 203-214. https://doi.org/10.1017/S0007485300031370
- Trapp S.C., Croteau R.B. 2001. Genomic organization of plant terpene synthases and molecular evolutionary implications. Genetics, 158: 811-832. https://doi.org/10.1093/genetics/158.2.811
- Van Dam M.H., Trautwein M., Spicer G.S., Esposito L. 2019. Advancing mite phylogenomics: Designing ultraconserved elements for Acari phylogeny. Mol Ecol Res, 19: 465-475. https://doi.org/10.1111/1755-0998.12962
- van der Hammen L. 1970. La segmentation primitive des Acariens. Acarologia, 12: 3-10.
- Van Zee J.P., Geraci N., Guerrero F., Wikel S., Stuart J., Nene V., Hill C. 2007. Tick genomics: the Ixodes genome project and beyond. Int J Parasitol, 37: 1297-1305. https://doi.org/10.1016/j.ijpara.2007.05.011
- Vieira F.G., Rozas J. 2011. Comparative genomics of the odorant-binding and chemosensory protein gene families across the Arthropoda: origin and evolutionary history of the chemosensory system. Genome Biol Evol, 3: 476-490. https://doi.org/10.1093/gbe/evr033
- Vurture G.W., Sedlazeck F.J., Nattestad M., Underwood C.J., Fang H., Gurtowski J., Schatz M.C. 2017. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics, 33: 2202-2204. https://doi.org/10.1093/bioinformatics/btx153
- Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One, 9: e112963. https://doi.org/10.1371/journal.pone.0112963
- Walter D.E., Proctor H.C. 1998. Feeding behaviour and phylogeny: observations on early derivative Acari. Exp Appl Acarol, 22: 39-50.
- Walter D.E., Proctor H.C. 1999. Mites: ecology, evolution, and behaviour. Amsterdam: Springer Netherlands. pp. 494.
- Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature, 420: 520-562. https://doi.org/10.1038/nature01262
- Weinstein S.B., Kuris A.M. 2016. Independent origins of parasitism in Animalia. Biol Lett, 12: 20160324. https://doi.org/10.1098/rsbl.2016.0324
- Woodring J. 1966. Color phototactic responses of an eyeless oribatid mite. Acarologia, 8: 382-388.
- Wrensch D.L., Kethley J.B., Norton R.A. 1994. Cytogenetics of holokinetic chromosomes and inverted meiosis: keys to the evolutionary success of mites, with generalizations on eukaryotes. In: Houck M.A., (Ed). Mites: Ecological and Evolutionary Analyses of Life-History Patterns. New York: Chapman & Hall. p. 282-343. https://doi.org/10.1007/978-1-4615-2389-5_11
- Wu C., Jordan M.D., Newcomb R.D., Gemmell N.J., Bank S., Meusemann K., Dearden P.K., Duncan E.J., Grosser S., Rutherford K. 2017. Analysis of the genome of the New Zealand giant collembolan (Holacanthella duospinosa) sheds light on hexapod evolution. BMC Genomics, 18: 795. https://doi.org/10.1186/s12864-017-4197-1
- Wybouw N., Pauchet Y., Heckel D.G., Van Leeuwen T. 2016. Horizontal gene transfer contributes to the evolution of arthropod herbivory. Genome Biol Evol, 8: 1785-1801. https://doi.org/10.1093/gbe/evw119
- Wybouw N., Van Leeuwen T., Dermauw W. 2018. A massive incorporation of microbial genes into the genome of Tetranychus urticae, a polyphagous arthropod herbivore. Insect Mol Biol, 27: 333-351. https://doi.org/10.1111/imb.12374
- Yu G., Wang L.-G., Han Y., He Q.-Y. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16: 284-287. https://doi.org/10.1089/omi.2011.0118
- Yunker C., Peter T., Norval R., Sonenshine D., Burridge M., Butler J. 1992. Olfactory responses of adult Amblyomma hebraeum and A. variegatum (Acari: Ixodiae) to attractant chemicals in laboratory tests. Exp Appl Acarol, 13: 295-301. https://doi.org/10.1007/BF01195086
- Zachvatkin A.A. 1941. Tyroglyphoidae (Acari). Moscow: Zoological Institute of the Acaemy of Science of the U.S.S.R.
- Zhang C., Rabiee M., Sayyari E., Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics, 19: 153. https://doi.org/10.1186/s12859-018-2129-y
- Zhou J., Wang C., Yoon S.-H., Jang H.-J., Choi E.-S., Kim S.-W. 2014. Engineering Escherichia coli for selective geraniol production with minimized endogenous dehydrogenation. J Biotech, 169: 42-50. https://doi.org/10.1016/j.jbiotec.2013.11.009
- Zinkler D. 1971. Vergleichende Untersuchungen zum Wirkungsspektrum der Carbohydrasen laubstreubewohnender Oribatiden. Zool Ges Verh: 149-153.



2022-03-21
Date accepted:
2022-05-12
Date published:
2022-06-08
Edited by:
Navajas, Maria

This work is licensed under a Creative Commons Attribution 4.0 International License
2022 Brückner, Adrian; Barnett, Austen A. ; Bhat, Prashant; Antoshechkin, Igor A. and Kitchen, Sheila A.
 Download article Download low definition
Download article Download low definitionDownload the citation
RIS with abstract
(Zotero, Endnote, Reference Manager, ProCite, RefWorks, Mendeley)
RIS without abstract
BIB
(Zotero, BibTeX)
TXT
(PubMed, Txt)