



Great molecular variation within the species Phytoseius finitimus (Acari: Phytoseiidae): implications for diagnosis decision within the mite family Phytoseiidae
Tixier, Marie-Stéphane1 ; Dos Santos Vicente, Victor2 ; Douin, Martial3 ; Duso, Carlo4 and Kreiter, Serge5
1✉ Montpellier SupAgro, UMR CBGP, INRA/ IRD/ CIRAD/ Montpellier SupAgro, 755 Avenue du Campus Agropolis, CS 30016, 34988 Montferrier-sur-Lez cedex, France
2Montpellier SupAgro, UMR CBGP, INRA/ IRD/ CIRAD/ Montpellier SupAgro, 755 Avenue du Campus Agropolis, CS 30016, 34988 Montferrier-sur-Lez cedex, France
3Montpellier SupAgro, UMR CBGP, INRA/ IRD/ CIRAD/ Montpellier SupAgro, 755 Avenue du Campus Agropolis, CS 30016, 34988 Montferrier-sur-Lez cedex, France
4Department of Agronomy, Food, Natural resources, Animals and Environment, University of Padova, Viale dell’Università 16, 35020 Legnaro (PD), Italy
5Montpellier SupAgro, UMR CBGP, INRA/ IRD/ CIRAD/ Montpellier SupAgro, 755 Avenue du Campus Agropolis, CS 30016, 34988 Montferrier-sur-Lez cedex, France
2017 - Volume: 57 Issue: 3 pages: 493-515
https://doi.org/10.24349/acarologia/20174168Keywords
Abstract
Molecular markers for differentiating microorganism, plant and animal species are currently used in taxonomy. The rapid technical developments (i.e. DNA extraction, PCR and Sanger sequencing) during these last decades as well barcoding approaches and associated conceptual definitions have led to the development of molecular diagnosis (i.e. Hebert et al. 2003a,b; 2004a,b; DeSalle et al. 2005; Goldstein and DeSalle 2011). Molecular markers were traditionally used when doubts on species identity arise from morphological comparisons and sometimes from biological features between populations. However, cryptic species are more and more revealed via comparison of DNA sequences of apparently morphologically similar specimens (i.e. Pons et al. 2006; Witt et al. 2006; Bickford et al. 2007; Valentini et al. 2009; Foottit et al. 2009; Pauls et al. 2010; Jorger et al. 2012; Kekkonen and Hebert, 2014; Kekkonen et al. 2015). The present study focuses on a species of the mite family Phytoseiidae: Phytoseius finitimus Ribaga. This family, containing 2,479 valid species (Demite et al. 2016), is one of the most well-known within the Mesostigmata, due to the predatory abilities of numerous species to control mite and small insect pests (McMurtry and Croft 1997; Gerson et al. 2003; McMurtry et al. 2013). Correct identification of these predators is clearly of huge importance for ensuring the success of biological control, as different species have different biological features (i.e. Tixier et al. 2006; McMurtry et al. 2013). Phytoseiidae species identification is essentially based on female morphological characters. However, the reliability of those characters as well as the low number of morphological features potentially available (due to the mite small size, 500 μm maximum in length) have led to the development of molecular studies to resolve questions concerning synonymies (i.e. Jeyaprakash and Hoy 2002; Noronha et al. 2003; Tixier et al. 2006, 2008a, 2010a, 2011b, 2012, 2014; Okassa et al. 2009, 2010, 2011, 2012; Kanouh et al. 2010a,b; Bowman and Hoy 2012; Chao et al. 2012; Navia et al. 2014; Rezende et al. 2015). For ensuring molecular diagnosis, a database containing DNA sequences associated to the correct species name is essential (i.e. DeSalle et al. 2005; Collins and Cruickshank 2013). When elaborating such a database for Phytoseiidae species, high molecular variations were observed within specimens identified as P. finitimus during samplings carried out in Italy and France, two areas where this species is most commonly found.
Phytoseius finitimus is a generalist predator and is quite common in Mediterranean vineyards (i.e. Ragusa and Ciulla 1991; Duso and Vettorazzo 1999; Duso and Fontana 2002; Peverieri et al. 2009; Miñarro and Kreiter 2012). Some studies have shown its ability to control phytophagous mites, whiteflies and thrips and to develop when fed with pollen (Pappas et al. 2013) and Ahmad et al. (2015) recently studied its interspecific competition features. The present study aims to elucidate the great molecular distances observed between four P. finitimus populations and to determine how these great variations are related or not to cryptic species occurrence. Answering this question is clearly important for biological control applications, as implementations and advice for agro-environmental management would be different if one or two species are named under P. finitimus. To determine if one or several species were present, an integrative taxonomic approach based on three molecular fragments and morphological analyses has been carried out. The number of analytical developments for assessing species within a DNA sequence dataset is increasing for ensuring diagnosis decision (i.e. Kekkonen et al. 2015). Traditional cluster tree-based and distance threshold-based approach as well as the overlapping ABGD (Automatic Barcode Gap Discovery) algorithm have been herein used to determine their respective performance and congruence. Furthermore, a compilation of genetic distances between Phytoseiidae species retrieved from already published works is also provided as a reference comparison point. This review aims to provide additional information that can be used in informing the decision and more broadly aims to assist further analyses on DNA molecular diagnosis within the family Phytoseiidae for delimiting intra- and interspecific frontiers.
Species and populations studied
Phytoseius finitimus, even if reported many times in the literature, is not naturally observed in great densities and is quite difficult to efficiently retrieve. It is known to be frequent in Italy and in Corsica (France), especially in vineyards (Kreiter et al. 2000; Duso and Fontana 2002; Demite et al. 2014). Four populations of P. finitimus were considered, with the double aims to study populations (i) from the two main areas where this species is most abundant (Italy and France) and (ii) from cultivated (vine and kiwi) and uncultivated areas (colonized by Viburnum lantana L.). Two populations were collected in Italy in the same locality on vines (Vitis vinifera L.) and on uncultivated V. lantana plants located besides the vine plot. Two populations were collected in France – Corsica, on vine and kiwi (Actinidia deliciosa C.F. Liang & A.R. Ferguson.) on plots separated by approximately 15 kms (Figure 1). The characteristics of the populations studied and accession numbers of DNA sequences in the Genbank database are presented in Table 1. The species Typhlodromus (Typhlodromus) phialatus Athias-Henriot, belonging to the sub-family Typhlodrominae, was used as an out-group for tree construction. Sequences of this latter species were obtained by present authors and already referenced in the Genbank database (12S rRNA: HM635274, ITSS: HQ404829, CytB mtDNA: HM635300) (Okassa et al. 2012; Tsolakis et al., 2012).


Molecular experiments
DNA extraction.
Total genomic DNA of a single female was extracted using a Qiagen DNeasy tissue kit (Qiagen, Hilden, Germany), according to the DNA extraction protocol « Purification of Total DNA from Animal Blood or Cells » (Spin-Column Protocol) adapted for extracting total DNA from mites (Kanouh et al. 2010b). Between six and thirteen females were considered per population (Table 1). After extraction, specimens were retrieved and mounted on slides according the method developed by Tixier et al. (2010 b).


DNA amplification and sequencing.
A nuclear ribosomal gene section including ITS1-5.8S-ITS2 (reported as ITSS) and two mitochondrial markers (12S rRNA, Cytochrome B mtDNA) were amplified within a multi-barcoding approach to avoid the current pitfall of barcoding using only one small DNA fragment (Moritz and Cicero 2004; Collins and Cruickshank 2013). These DNA fragments have been used for species diagnosis within Phytoseiidae (Jeyaprakash and Hoy 2002; Tixier et al. 2006, 2008a, 2010a, 2011b, 2012, 2014; Okassa et al. 2009, 2010, 2011, 2012; Kanouh et al. 2010a,b; Navia et al. 2014; Rezende et al. 2015). Primers and thermal cycling are those reported in Tixier et al. (2012). The PCR reactions were performed in a 25 µL volume, containing 4 µL of mite DNA for mt markers and 2 µL for ITSS, 2.5 µL (1 mM) of buffer 10X, 1 µL (1.5 mM) of MgCl2, 0.5 µL (0.05 mM for each) DNTPs, 0.175 µL (0.7 μm) for each primer, 0.125 µL (0.625 U) of Taq Qiagen and 16.525 µL of water. Electrophoresis was carried out on a 1.5 % agarose gel in 0.5 X TBE buffer during 20 min at 135 volts. PCR products were purified using ExoSAP-IT (Amersham) and sequenced along both strands using Dynamic ET Terminator Cycle Sequencing kit. The sequencer used was the Megabase 1,000 apparatus. Sequences were aligned and analysed with Geneious v3.5.4 (Drummond et al. 2007).
Data analyses.
A preliminary analysis was conducted on the coding sequences (CytB mtDNA) to check for the absence of stop codons. Three molecular diagnosis approaches were used to assess the occurrence of cryptic species within the four populations of P. finitimus.
Tree-based cluster approach.
Neighbour-joining trees were constructed using the Kimura 2-parameter model and node support was determined using 1,000 bootstrap replicates calculated in Mega 6.0.6® (Tamura et al. 2013). The K2P model was used to make our results comparable with other studies on Phytoseiidae. Furthermore to determine how other tree models perform for resolving the question on cryptic species, Parsimony and Bayesian analyses were carried out for each fragment and combined dataset (applying simple partition procedure with one partition corresponding to each DNA marker). As for some specimens, it was impossible to obtain PCR products (especially for ITS), the combined data set was analysed following Wiens (1998a,b), Wiens et al. (2005) and Zheng and Wiens (2016), who demonstrated that missing data do not affect the phylogenetic accuracy.
The Incongruence Length Difference (ILD) P value (P= 0.07) suggests no conflicting signals between markers (Mickevich and Farris 1981; Sullivan 1996; Farris et al. 1994; Cunningham 1997).For Parsimony analyses a heuristic search procedure repeated 1,000 times, was performed with randomized taxa additions and branch-swapping algorithm (TBR). To reduce misleading effects of homoplasious characters, an a posteriori re-weighting was applied according to the rescaled consistency index (RC) after each tree search, until the number of trees stabilised (Farris 1969, 1989) (PAUP*, v.4.0b.10 / Swofford, 2002). Bayesian analysis was carried out using MrBayes 3.1 (Ronquist and Huelsenbeck, 2003); the best-fit-substitution models were determined by Modeltest 3.07 in PAUP (Posada and Crandall 1998) through hierarchical likelihood-ratio tests (12S rRNA: HKY+G; CytB mtDNA: TIM+I; ITSS: HKY+G). The number of categories used to approximate the gamma distribution was set at four, and four Markov chains were run for 100,0000 generations. Stabilisation of model parameters (burn-in) occurred around 250 generations.
As tree clades do not necessary reflect species clusters but can represent populations of a same species (i.e. Moritz and Cicero 2004; Collins and Cruickshank 2013), the classical threshold-based approach was applied and distance matrices (using the Kimura 2-parameter) model were elaborated. With this approach, ideally no overlap should occur between intra and interspecific distances (i.e. Meyer and Paulay 2005). Even if the no-overlap model is sometimes point of contention especially because of sampling completion, knowledge of species considered and associated taxonomical features, distance comparison is currently used to distinguish between species for practical assignments. Two approaches for comparing distances were applied.
The overlap distance criteria.
First, distances within and between the four populations were manually compared to determine gap occurrence. In addition, the ABGD (Automatic Barcode Gap Discovery) algorithm was applied. It aims at automatically determining the occurrence of different species within a dataset based on gap between batches of sequences pairs (Puillandre et al. 2012a), detecting the first barcode gap and using it to partition the dataset into candidate species. Analyses were performed in November 2016 on the web interface (http://wwwabi.snv.jussieu.fr/public/abgd/). They were conducted using three metrics (Jukes-Cantor (JC), Kimura 2 parameter (K2P) and simple p-distances) and default parameter values.
Empirical comparisons to references.
Distances usually constitute a useful point of comparison when the taxonomic status of the taxa compared is sure (Robinson et al. 2009). Thus, the genetic distances herein obtained were compared to those mentioned in 15 publications for 24 Phytoseiidae species. Only distances issued from published works and not calculated from all Phytoseiidae sequences deposited in Genbank were considered, because of great incertitude on the true identity of the sequences deposited in this world database (i.e. Tixier et al. 2011a). Two distances were compiled: (i) one corresponding to the maximal value observed within a same species (maximal intraspecific variation) and (ii) one corresponding the minimal value observed between two species of a same genus [minimal interspecific variation assumed to be the "Nearest Neighbour" (Kekkonen et al. 2015)].
Morphological analyses
Characters considered.
As female dorsal seta lengths are currently used to discriminate between Phytoseiidae species (Chant and McMurtry 2007), the seventeen dorsal idiosomal setae were measured for 5 to 10 females issued from the same populations as sequenced specimens: j1, j3, j4, j5, j6, J2, J5, z2, z3, z5, z4, Z4, Z5, s4, s6, r3 and R1. The lengths of the seta JV5 and the macroseta on the basitarsus of the leg IV (St IV) were also considered. Finally, peritreme, spermatheca, ventrianal shield and chelicera shapes were observed. Terminology for seta notation follows that of Lindquist and Evans (1965) as adapted by Rowell et al. (1978) for Phytoseiidae.
Data analyses. The data corresponding to the 17 continuous characters were normally distributed and variances were equal (Bartlett test). ANOVA analyses were performed for each character to determine differences between the four populations. A principal component analysis was also carried out to determine whether the combination of the morphological characters differentiates specimens considered. All statistical analyses were carried out with Statistica (Statsoft France 2010). To determine the variation of each seta length, the rCL95 corresponding to the percentage of variation around the mean was calculated according to the formula provided in Tixier (2012). Furthermore, min and max values of the interval assumed to include 95 % of specimens of a same species for each setal length were modelled using the abacus provided in Tixier (2012) to determine whether the observed variation would be included in the modelled "normal" intraspecific variation.


Molecular analyses.
437, 418 and 690 base pairs (bp) were aligned for the 12S rRNA, Cytb mtDNA, and ITSS genes, respectively. Quite similar and constant rates of nucleotide substitutions were observed for all the populations studied. A BLAST search in Genbank showed that the sequences aligned with those of Phytoseiidae. The overall genetic distances between the specimens of P. finitimus for 12S rRNA (mean = 2.9 %, min-max = 0-9 %) and CytB mtDNA (mean = 7.6 %, min-max = 0-23 %) are high in regards to those obtained in other studies (Table 3) questioning the occurrence of cryptic species.


Diagnosis decision and tree cluster criteria.
The same tree topologies were observed with the three algorithms (Neighbour Joining, Parsimony, Bayesian analyses) for the three markers considered. Three clades were observed on the trees obtained with the two mitochondrial markers (12S rRNA, CytB mtDNA): one containing specimens collected in Corsica on vine, one containing specimens collected in Italy on vine and in Corsica on kiwi and one containing specimens collected in Italy on V. lantana (Figures 2, 3). One specimen collected on vine in Corsica was however located in the clade containing specimens from vine-Italy and kiwi-Corsica. These clades could be three species or three populations of a same species. Specimens included in the two former clades are more closer to each other than to specimens included in the third. Two not well-sustained clades are observed on the ITSS tree (Figure 4), one containing specimens from V. lantana and one containing the specimens of the three other populations. Furthermore, the same clades as those obtained with mitochondrial markers were also obtained when concatenated data were considered for both Parsimony and Bayesian analyses (Figure 5).

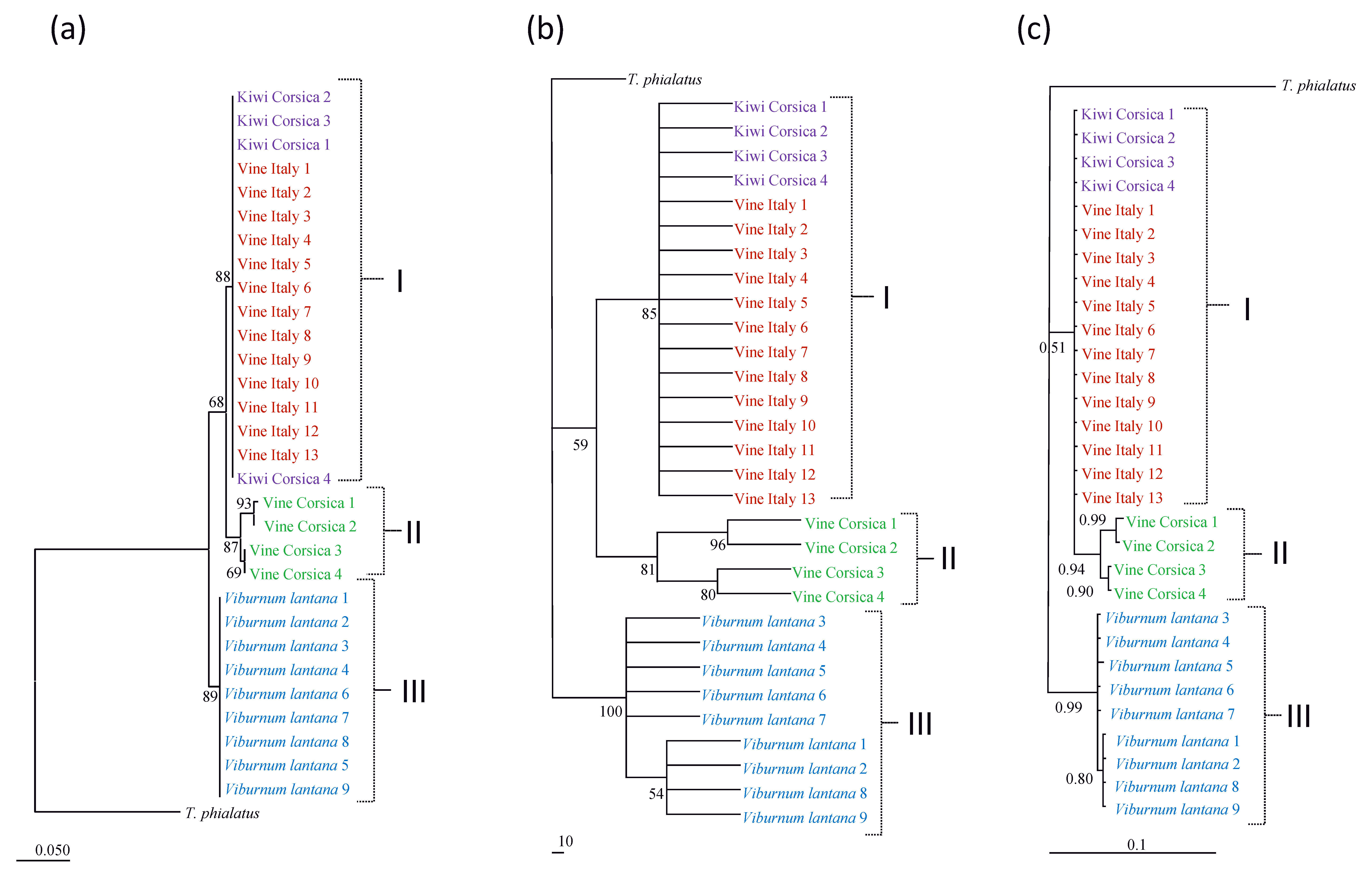




With the ABGD analyses, the same groups were obtained for the Jukes-Cantor (JC69) and Kimura (K80) TS/TV distances but different clusters were observed for the P distance (Table 3). For the 12S rRNA and CytB mtDNA markers, four and five groups were observed with the K80 and JC69 distances, respectively. Globally, the same groups as those observed on the phylogenetic trees were obtained, but more clustering was observed within the population collected on vine in Corsica (two clusters for 12S rRNA and three clusters for CytB mtDNA). Using P distance, only two groups were observed with the two mitochondrial markers, one containing specimens collected on V. lantana and one containing all the others. For the ITSS marker, one group was observed whatever the distance considered. The ABGD method provides thus different information and groups depending on (i) mitochondrial DNA vs nuclear markers and (ii) the distance considered.
Diagnosis decision and comparison to references in literature. The 12S molecular distances between clades I and II (0-5 %) correspond to values currently retrieved in literature for specimens belonging to the same species (Table 4). Furthermore, such distances are lower than the minimal distances observed until now between species of a same genus. In one case corresponding to distances between Phytoseiulus persimilis Athias-Henriot and P. macropilis (Banks), the presently observed distances are higher than the minimal distance observed between these two species (4 %). The maximal 12S rRNA distances between V. lantana specimens and specimens of clades I and II (9 %) is higher than the maximal intraspecific distances observed until now within a Phytoseiidae species (8 %) (Tables 2, 4). This value even if close to minimal distances observed between Neoseiulus californicus and N. fallacis (Garman) [10 % in Tixier et al. (2014) and 9.5 % in Jeyaprakash and Hoy (2002)] is however lower than distances observed until now between two Phytoseiidae species of the same genus.

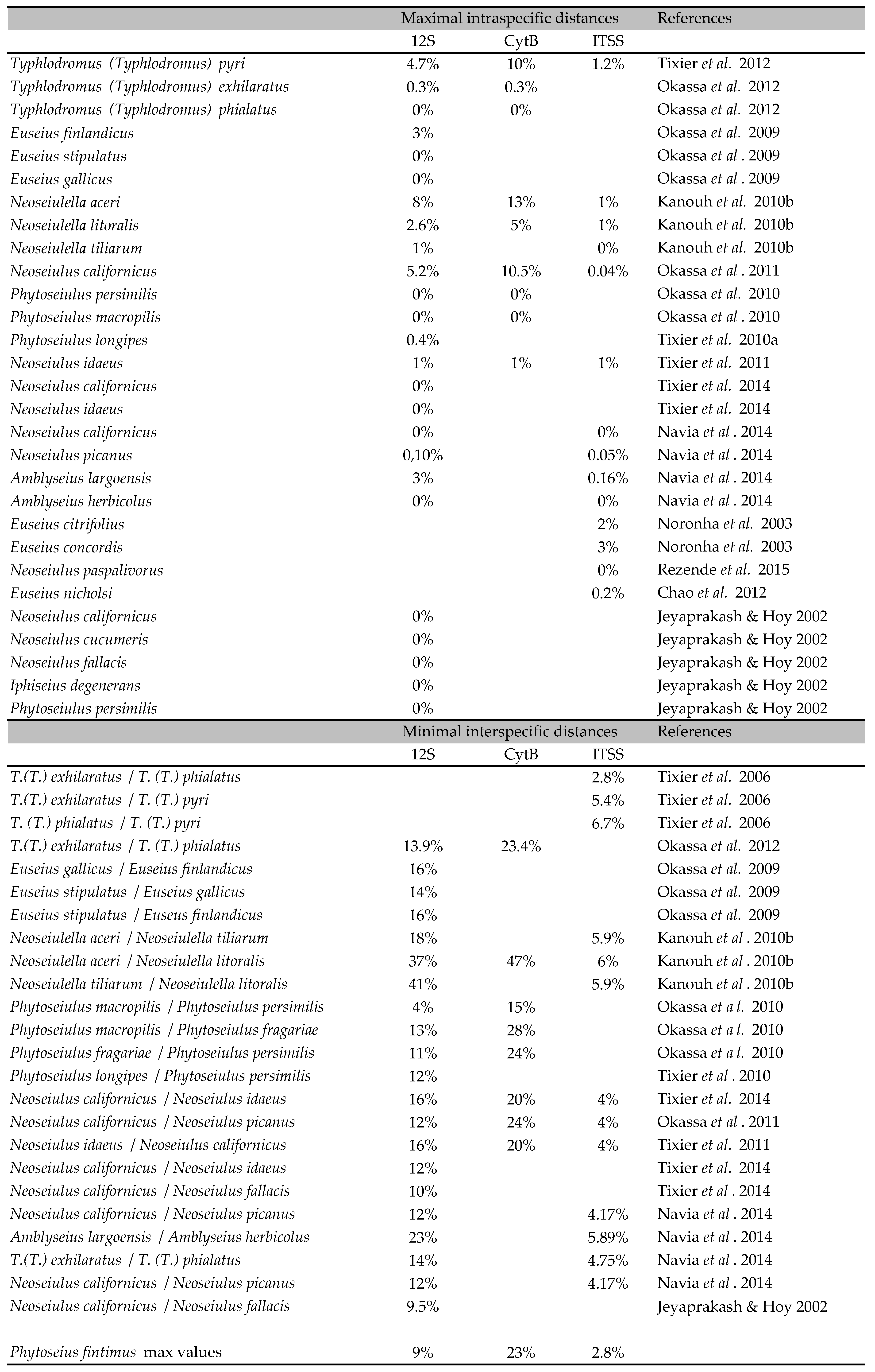
The mean CytB distances between specimens from clades I and II are usually lower than genetic distances observed within a same Phytoseiidae species (Tables 2, 4). However, maximal distances (16 %) observed between specimens from vine in Italy and France, and kiwi from France as well those within vine-Corsica population (13 %), are higher or equal to the maximal intraspecific distances observed until now for Phytoseiidae (13 % for N. aceri). The maximal CytB distances between specimens from V. lantana and the three other populations (23%) are higher than those already observed within a same Phytoseiidae species. This value is just below the minimal interspecific distances observed between species of some genus (23.4 % between T. (T.) phialatus and T. (T.) exhilaratus and 24 % between N. californicus and N. picanus in Okassa et al. (2011, 2012)).
The highest ITSS distances are observed between specimens collected on V. lantana and the specimens of the other populations. The distances between clades I and II are very low and included in the variation range of intraspecific distances retrieved in literature (Table 3). The mean distances between these two clades and specimens from V. lantana are usually lower than those observed between different species of a same Phytoseiidae genus. The maximal distance is however similar to the distances obtained between T. ( T.) phialatus and T. (T.) exhilaratus (two morphological close species) by Tixier et al. (2006) whereas Navia et al. (2014) reported a distance of 4.75 % between these two same latter species (Table 4).
Morphological analyses.
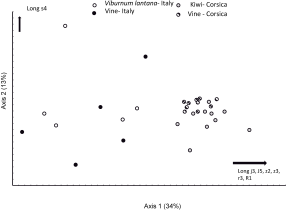
Some statistical differences are observed for twelve continuous characters (Table 5). However, the statistical groups show that measurements of specimens from V. lantana are usually not different from specimens from the other three populations. The most frequent significant differences are observed between specimens from Corsica (vine and kiwi) and from Italy (vine and V. lantana). On the two axes of the multifactorial analysis explaining 47 % of the variation (Figure 7), specimens collected on V. lantana are close to those collected on vine-Italy, these two populations being quite well separated from the specimens collected in Corsica, both on vine and kiwi. The variation within and between populations from Italy is lower than that observed within and between populations from Corsica. Mean differences in seta length even if statically significant are however usually low. The most important difference is observed in s6 length, this setae being significantly shorter for specimens collected on V. lantana (mean = 81 μm) than for specimens of the three other populations (88, 89, and 94 μm for mites collected on vine in Italy, on kiwi in Corsica and on vine in Corsica, respectively).
egintable*[htbp!]% table5 entering ncludegraphics[width=argeurfigurehorizontale]../Articles/00429-Tixier/Table5.pdfaptionMorphological measurements of the characters considered (μm) (mean, Standard deviation in parentheses, min-max values) for the four populations considered (kiwi in Corsica, Viburnum lantana in Italy, vine in Italy and vine in Corsica) and significance of between-population comparison. rCL95 values (%) and min-max interval modelled according to Tixier (2012). StIV: macroseta lengths on the basitarsus of the leg IV.ndtable*

However the absolute value between these seta lengths, ranging between 7 and 13 μm, is according to the modelling of intraspecific seta length range proposed by Tixier (2012, 2013) included in intraspecific variation (Table 4). The rCL95 calculated according to Tixier (2012) shows that most setae have a variation around the mean lower than 20 %, except for the smallest setae. This result is in accordance with results of Tixier (2012) when modelling the intraspecific variation rate around the mean for setae length of Phytoseiidae dorsal shield. These modelled variation intervals around the means (Tixier 2012) include the variation range of specimens herein studied for all seta considered.
Furthermore observations of shapes of spermatheca (Figure 8) as well as ventrianal shield chaetotaxy and shape and peritremal extremity (reaching z2) do not reveal differences between the populations considered.
This study shows (i) the difficulty in interpreting DNA variation for species diagnosis and (ii) the necessity of an integrative taxonomic approach. Clear conclusion can be proposed for two clades (clades I and II) among the three observed on the mitochondrial DNA trees. Because (i) these two clades were not separated using the nuclear ITSS markers, (ii) distance overlap was observed for all markers (nuclear and mitochondrial) between intra and inter-population distances, (iii) these populations belong to a same group with the ABGD method (using P distance), one specimen of vine-Corsica is included in the clade containing specimens from vine-Italy and kiwi-Corsica and (iv) the higher inter-population distances were much lower than the already observed ones between two species of the same genus, the convergent decision was that these two clades belong to the same species. However, for the status of the third clade containing specimens collected on V. lantana, the decision was more difficult. Because of (i) the well-separated clade including those specimens with both mitochondrial and nuclear markers, (ii) the high distances between those specimens and specimens from the three other populations, (iii) no clear overlap between sequences of these two groups for the 12S rRNA fragment and (iv) results obtained based on mtDNA fragments with the ABGD approach, two hypotheses could be proposed: (i) specimens of the four populations considered belong to a same species characterised by high molecular polymorphisms, or (ii) specimens from V. lantana belong to a species very close to the specimens of the three other populations. The final decision was made based on the observation of morphological characters. No difference between seta lengths of the four populations is observed, suggesting that they belong to the same species. According to the markers and analytic concepts considered, diagnosis conclusion can be different and decision hard to dress.
Molecular markers and pitfalls.
Mitochondrial markers showed much more variation than was observed with the nuclear marker. ITSS and mitochondrial markers provide thus different information; it is not possible based only on trees to give a clear decision on the state of clades obtained (different species or populations). Such a result was observed for other Phytoseiidae species for example indicating possible cryptic species within Typhlodromus (Typhlodromus) pyri Scheuten, Neoseiulella aceri and Neoseiulus californicus (Kanouh et al. 2010a; Okassa et al. 2011; Tixier et al. 2012) as well for other groups of mites in Mesostigmata (Roy et al. 2010), Ixodidae (Leo et al. 2010); Hydrachnidia (Stalstedt et al. 2013), Oribatida (Rosenberger et al. 2013; Kreipe et al. 2015), Tetranychidae (Navajas and Boursot 2003; Ros et al. 2008), Eriophyidae (Skoracka and Dabert 2010), especially for the COI mtDNA marker.
Such mitochondrial polymorphism can be due to the nature of molecule evolution (i.e. mtDNA is more sensitive to population bottlenecks, the absence of recombination in mtDNA) and to ecological features (i.e. dispersal, fecundity) that lead to population separations. Furthermore, Phytoseiidae mites are haplodiploid. Even if there are few studies on the impact of such features on mt DNA variation outside of Hymenoptera, Lohse and Ross (2015) suggest that this characteristic can imply a higher introgressions for mitochondrial compared to nuclear markers ; they report current incongruence between nuclear and mitochondrial trees in haploidiploid organisms, including spiders.
As a consequence, some artificial groups can be obtained, underlying the necessity to use an integrative taxonomical approach, combining morphological and molecular characters as well as different DNA fragments and associated conceptual analyses (i.e. Dayrat 2005).
Which analytic methods to use?
This study first stresses the difficult in interpreting phylogenetic trees. The same clades were obtained for the three algorithms used (Neighbour-Joining, Parsimony and Bayesian analyses), suggesting that at low taxonomic level the Neighbour-Joining tree provides the same clustering as the other model-based analyses (Holder and Lewis 2003; Yang and Rannala 2012). In addition the concatenated analysis of the three DNA markers does not provide a clearer answer as the same clusters were obtained as those with the two mitochondrial markers. The concatenated tree thus can hide some information as the signal obtained clearly depends on the resolution level of each DNA marker considered.
This study also stresses the necessity of having clear distance references for comparison. The ABGD aims to construct its own rules based on a data set (without a priori hypothesis on taxon status) (Puillandre et al. 2012a). This method overcomes common threshold applications, reported to be variable according to taxa considered (i.e. Collins and Cruickshank 2013). For the present dataset, more than one species are emphasized using the two mitochondrial markers. Four and five groups are even observed using the JC69 and K80 distances, suggesting (i) the high sensitivity of this clustering method and (ii) the impact of the number of sequences analysed (Puillandre et al. 2012b). Furthermore, even with the ABGD analysis based on P distance, the specimens from V. lantana were always assumed to belong to a different species. However using the ITSS marker, the results converge with other analyses, especially morphology suggesting that all specimens belong to a same species. The number of populations and sequences were quite low which can lead to artificial groups significantly detected by ABGD approach (Lhose 2009; Puillandre et al. 2012b). To our knowledge, this latter method has never been applied for the markers 12S rRNA, CytB mtDNA and only one time for ITS2 (Schwarzfeld and Sperling 2015); it has been essentially used for the traditional barcode fragment (COI mtDNA) and in some rare cases for 16S rRNA (i.e. Puillandre et al. 2012b; Jörger et al. 2012; Guarnizo et al. 2015; Han et al. 2016). Results herein obtained can be due to the fact that other DNA fragments were considered and prior parameters not adapted. Furthermore, it was also carried out only once on mites, on feather mites using COI (Doña et al. 2015) making conclusions of ABGD relevance for mite species delimitation difficult.
This study contributes to the characterization of decision rules for molecular diagnoses within Phytoseiidae and more generally speaking within mites. It is the first time that molecular variation of a species of the subfamily Phytoseiinae is studied. Its variation is higher that already observed within species of the other two subfamilies (Typhlodrominae and Amblyseiinae). Testing additional species of the sub-family Phytoseiinae would be interesting for understanding how subfamily and associated biological/ecological features can impact intraspecific variations (i.e. low dispersal, small population size). The present genetic distances are the highest ever observed within a Phytoseiidae species. Yet, it seems that genetic distances higher than 9 %, 23 % and 2.8 % would correspond to interspecific distances for the markers 12S rRNA, CytB mtDNA and ITSS, respectively. Those results are quite difficult to compare with those obtained for other groups, as the threshold can be different according to taxonomic entities (i.e. Collins and Cruickshank 2013). Furthermore, CytB mt DNA and 12S rRNA markers have been rarely used for species diagnosis within other mite groups. A recent study on ticks using the 12S rRNA markers proposes a minimal interspecific distance of 13.2% and an average intraspecific distance of 1.8%; no data is provided on maximal intraspecific distance (Lv et al. 2014).
These simple comparisons need to be completed with statistical analyses to determine the validity (and associated error) of a barcoding decision depending on the distance obtained between two sequences and to their proximity with the lower limit of intraspecific variation. Furthermore, when comparing with literature, these rules seem to be different depending on the genus as minimal genetic distance of 4 % for the 12S rRNA is observed between two close species (P. persimilis and P. macropilis). However, in this latter case, it is worthwhile to test if: (i) this low intraspecific distance is due to particular biological features of species of the atypical genus Phytoseiulus within Phytoseiidae, or (ii) P. macropilis and P. persimilis are synonyms and a 12S rRNA distance of 4 % would correspond to an intraspecific value, as for all the Phytoseiidae species tested until now.
Implications for biological control?
Finally, for biological control matters, further experiments should be planned to determine whether the present mitochondrial clades show different behaviour and biological features. Tixier et al. (2010a) observed that mitochondrial clades within the species Phytoseiulus longipes Evans were associated with different feeding abilities (prey preferences). The closest populations with molecular markers correspond to specimens collected in cultivated areas (vine-Italy and -Corsica and kiwi-Corsica) but in geographically distinct areas. The well-differentiated population was collected from uncultivated plants (V. lantana) nearby the vine-Italy plot. It is interesting to note that morphological analyses do not show the same similarities, as morphological similarity seems more associated with geographical distances than with plants and cultivated situation. Further investigations would be to determine how selection pressures associated with agricultural practices are linked to such a differentiation for assessing the consequences on colonisation of specimens of P. finitimus dispersing from uncultivated vegetation towards crops.
We are grateful to the three anonymous reviewers and their valuable comments that allow article improvement. We also thank Andrea Baldassar an Italian Master student that worked on this project.
Ahmad S., Pozzebon A., Duso, C. 2015 — Predation on heterospecific larvae by adult females of Kampimodromus aberrans, Amblyseius andersoni, Typhlodromus pyri and Phytoseius finitimus (Acari: Phytoseiidae) — Exp. Appl. Acarol., 67: 1-20.
Bickford D., Lohman D.J., Sohdi N.S., Ng P.K., Meier R., Winker K., Ingram K.K., Das I. 2007 — Cryptic species as a window on diversity and conservation — Trend. Ecol. Evol. 22(3): 148-155.
Bowman H.M., Hoy M.A. 2012 — Molecular discrimination of phytoseiids associated with the red palm mite Raoiella indica (Acari: Tenuipalpidae) from Mauritius and South Florida — Exp. Appl. Acarol. 57(3-4): 395-407. doi:10.1007/s10493-012-9549-6 ![]()
Chant D.A., McMurtry J.A. 2007 — Illustrated keys and diagnoses for the genera and subgenera of the Phytoseiidae of the world (Acari: Mesostigmata) — West Bloomfield, MI: Indira Publishing House, 220 pp.
Chao Y., YuanXi L., XianMing Y., JingTao S., XueNong X., XiaoYue H. 2012 — Genetic variation among natural populations of Euseius nicholsi (Acari: Phytoseiidae) from China detected using mitochondrial coxI and nuclear rDNA ITS sequences — Syst. Appl. Acarol. 17(2): 171-181.
Collins R.A., Cruickshank R.H. 2013 — The seven deadly sins of DNA barcoding — Mol. Ecol. Resources 13 (6): 969-975.
Cunningham C.W. 1997 — Can three incongruence tests predict when data should be combined? — Mol. Biol. Evol. 14: 733-740. doi:10.1093/oxfordjournals.molbev.a025813 ![]()
Dayrat B. 2005 — Towards integrative taxonomy — Biol. J. Linn. Soc. 85: 407-415. doi:10.1111/j.1095-8312.2005.00503.x ![]()
Demite P.R., Moraes G.J. de, McMurtry J.A., Denmark H.A., Castilho R. C. 2016 — Phytoseiidae Database — Available from: www.lea.esalq.usp.br/phytoseiidae (accessed 04/11/2016)
DeSalle R., Egan M.G., Siddall M. 2005 — The unholy trinity: taxonomy, species delimitation and DNA barcoding — Philos. Trans. R. Soc. B-Biol. Sci. 360: 1905-1916.
Do-a J., Diaz-Real J., Mironov S., Bazaga P., Serrano D., Jovani R. 2015 — DNA barcoding and minibarcoding as a powerful tool for feather mite studies — Mol. Ecol. Res. 15(5): 1216-25. doi:10.1111/1755-0998.12384 ![]()
Duso C., Vettorazzo E. 1999 — Mite population dynamics on different grape varieties with or without phytoseiids released (Acari: Phytoseiidae) — Exp. Appl. Acarol. 23: 741-763. doi:10.1023/A:1006297225577 ![]()
Duso C., Fontana P. 2002 — On the identity of Phytoseius plumifer (Canestrini & Fanzago, 1876) (Acari: Phytoseiidae) — Acarologia 42: 127-136.
Drummond A.J., Ashton B., Cheung M., Heled J., Kearse M., Moir R., Stones-Havas S., Thierer T., Wilson A. 2007 — Geneious v3.5. Available from http://www.geneious.com/. Accessed July 2016.
Farris J.S. 1969 — A successive approximations approach to character weighting — Syst. Zool. 18: 374-385. doi:10.2307/2412182 ![]()
Farris J.S. 1989 — The retention index and the rescaled consistency index — Cladistics 5: 417-419.
Farris J.S., Kallersjo M., Kluge A.G., Bult C. 1994 — Testing significance of incongruence — Cladistics 10: 315-319.
Foottit R.G., Maw H.E.L, Havill N.P, Ahern R.G., Montgomery M.E 2009 — DNA barcodes to identify species and explore diversity in the Adelgidae (Insecta: Hemiptera: Aphidoidea) — Mol. Ecol. Resources 9: 188-195. doi:10.1111/j.1755-0998.2009.02644.x ![]()
Gerson U., Smiley R.L., Ochoa T. 2003 — Mites (Acari) for pest control — Blackwell Science, Oxford, United Kingdom. 539 pp.
Goldstein P.Z., DeSalle R. 2011 — Integrating DNA barcode data and taxonomic practice: determination, discovery, and description — Bioessays 33: 135-147.
Guarnizo C.E., Paz A., Munoz-Ortiz A., Flechas S.V., Mendez-Narvaez J., Crawford A.J. 2015 — Barcoding survey of Anurans across the Eastern Cordillera of Colombia and the impact of the Andes on cryptic diversity — PLoS One 10(5): e0127312. doi:10.1371/journal.pone.0127312 ![]()
Han T., Lee W., Lee S., Park G., Park H. 2016 — Reassessment of species diversity of the subfamily Denticollinae (Coleoptera: Elateridae) through DNA Barcoding — PloS One 11(2): e0148602. doi:10.1371/journal.pone.0148602 ![]()
Hebert P.D.N., Cywinska A., Ball S.L., DeWaard J.R. 2003a — Biological identifications through DNA barcodes — Proc. R. Soc. London Ser. B-Biol. Sci. 270: 313-321.
Hebert P.D.N., Ratnasingham S., deWaard J.R. 2003b — Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species — Proc. R. Soc. London Ser. B-Biol. Sci. 270: S96-S99.
Hebert P.D.N., Penton E.H., Burns J.M., Janzen D.H., Hallwachs W. 2004a — Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator — Proc. Nat. Acad. Sci. USA 101: 14812-14817.
doi:10.1073/pnas.0406166101Hebert P.D.N., Stoeckle M.Y., Zemlak T.S., Francis C.M. 2004b — Identification of birds through DNA barcodes — Plos Biology 2: 1657-1663. doi:10.1371/journal.pbio.0020312 ![]()
Holder M., Lewis P.O. 2003 — Phylogeny estimation: traditional and Bayesian approaches — Nature reviews genetics 4 : 275-284.
Jeyaprakash A., Hoy M.A. 2002 — Mitochondrial 12S rRNA sequences used to design a molecular ladder assay to identify six commercially available phytoseiids (Acari: Phytoseiidae) — Biol. Contr. 25: 136-142. doi:10.1016/S1049-9644(02)00056-7 ![]()
Jorger K.M., Norenburg J.L., Wilson N.G., Schrödl M. 2012 — Barcoding against a paradox? Combined molecular species delineations reveal multiple cryptic lineages in elusive meiofaunal sea slugs — BMC Evolution. Biol. 12: 245.
Kanouh M., Tixier M.-S., Guichou S., Cheval B., Kreiter S. 2010a — Two synonymy cases within the genus Neoseiulella (Acari: Phytoseiidae): is the molecular evidence so evident? Biol. J. Linn. Soc. 101: 323-344.
Kanouh M., Tixier M.-S., Okassa M., Kreiter S. 2010b — Phylogenetic and biogeographic analysis of the genus Phytoseiulus (Acari: Phytoseiidae) — Zool. Scr. 39: 450-461.
Kekkonen M., Hebert P.D.N. 2014 — DNA barcode-based delineation of putative species: Efficient start for taxonomic workflows — Mol. Ecol. Resources 14: 706-715.
doi:10.1111/1755-0998.12233Kekkonen M., Mutanen M., Kaila L., Nieminen M., Hebert P.D.N. 2015 — Delineating species with DNA Barcodes: A case of taxon dependent method performance in moths — PLoS One 10(4): e0122481.
doi:10.1371/journal.pone.0122481Kreipe V., Corral-Hernandez E., Scheu S., Schaefer I., Maraun M. 2015 — Phylogeny and species delineation in European species of the genus Steganacarus (Acari, Oribatida) using mitochondrial and nuclear markers — Exp. Appl. Acarol. 66(2): 173-86.
Kreiter S., Tixier M.-S., Auger P., Muckenstrum N., Sentenac G., Doublet B., Weber M. 2000 — Phytoseiid mites of vineyards in France — Acarologia 41: 75-94.
Leo S. S., Pybus M.J., Sperling F.A.H. 2010 — Deep mitochondrial DNA lineage divergences within Alberta populations of Dermacentor albipictus (Acari: Ixodidae) do not indicate distinct species — J Med. Entomol. 47: 565-574. doi:10.1093/jmedent/47.4.565 ![]()
Lindquist E.E., Evans G.W. 1965 — Taxonomic concepts in the Ascidae, with a modified setal nomenclature for the idiosoma of the Gamasina (Acarina: Mesostigmata) — Mem. Entomol. Soc. Canada 47: 1-64. doi:10.4039/entm9747fv ![]()
Lohse K. 2009 — Can mtDNA barcodes be used to delimit species? A response to Pons et al. (2006) — Syst. Biol. 58: 439-442. doi:10.1093/sysbio/syp039 ![]()
Lohse K., Ross L. 2015 — What haplodiploids can teach us about hybridization and speciation — Mol. Ecol. 24 : 5075-5077. doi:10.1111/mec.13393 ![]()
McMurtry J.A., Croft B.A 1997 — Life-styles of phytoseiid mites and their roles in biological control — Ann. Rev. Entomol. 42: 291-321.
McMurtry J.A., Moraes G.J. de, Sourasso N.F. 2013 — Revision of the life styles of phytoseiid mites (Acari: Phytoseiidae) and implications for biological control strategies — Syst. Appl. Acarol. 18: 297-320.
Meyer, C.P. & Paulay, G. (2005). DNA Barcoding: Error rates based on comprehensive sampling. PLoS Biology 3(12): e422. doi:10.1371/journal.pbio.0030422 ![]()
Mickevich M.F., Farris F.J. 1981 — The implications of congruence in Menidia — Syst. Zool. 30: 351-369. doi:10.2307/2413255 ![]()
Mi-arro M., Kreiter S 2012 — Fitoseidos em los vi-edos de la denominación Vino de Calidad de Cangas (Asturias) — Bol. San. Veg. Plagas 38: 73-82.
Moritz C., Cicero C. 2004 — DNA barcoding: promise and pitfalls — PLoS Biology, 2 (10): e354-10.1371.
Navajas M., Boursot P. 2003 — Nuclear ribosomal DNA monophyly versus mitochondrial DNA polyphyly in two closely related mite species: the influence of life history and molecular drive — Proc. Royal Soc. London B: Biol. Sci., 270 (Suppl 1) : S124-S127. doi:10.1098/rsbl.2003.0034 ![]()
Navia D., Domingos C.A., Mendonça R.S., Ferragut F., Rodrigues M.A.N, de Morais E.G.F., Tixier M.-S., Gondim Jr. M.G.C. 2014 — Reproductive compatibility and genetic and morphometric variability among populations of the predatory mite, Amblyseius largoensis (Acari: Phytoseiidae), from Indian Ocean islands and the Americas — Biol. Contr. 72: 17-29.
Noronha A.C.S., Mota A., Moraes G.J., Coutinho L.L. 2003 — Molecular characterization of mite populations of Euseius citrifolius Denmark & Muma and Euseius concordis (Chant) (Acari: Phytoseiidae) using sequences of the ITS1 and ITS2 regions — Neotrop. Entomol. 32: 591-596.
Okassa M., Kreiter S., Guichou S., Tixier M.-S. 2011 — Molecular and morphological boundaries of the predator Neoseiulus californicus McGregor (Acari: Phytoseiidae) —Biol. J. Linn. Soc. 104: 393-406.
Okassa M., Kreiter S., Tixier M.-S. 2012 — Molecular identification of all life stages of a mite species (Mesostigmata: Phytoseiidae): Typhlodromus (Typhlodromus) exhilaratus — Exp. Appl. Acarol. 57: 105-116.
Okassa M., Tixier M.-S., Cheval B., Kreiter S. 2009 — Molecular and morphological evidence for new species status within the genus Euseius (Acari: Phytoseiidae) — Can. J. Zool. 87: 689-698.
Okassa M., Tixier M.-S., Kreiter S. 2010 — Morphological and molecular diagnostics of Phytoseiulus persimilis and Phytoseiulus macropilis (Acari: Phytoseiidae) — Exp. Appl. Acarol. 52(3): 291-303.
Pappas M.L., Xanthis C., Samaras K., Koveos D.S., Broufas G.D. 2013 — Potential of the predatory mite Phytoseius finitimus (Acari: Phytoseiidae) to feed and reproduce on greenhouse pests — Exp. Appl. Acarol. 61(4): 387-401.
Pauls S.U., Blahnik R.J., Zhou X., Wardwell C.T., Holzenthal R.W. 2010 — DNA barcode data confirm new species and reveal cryptic diversity in Chilean Smicridea (Smicridea) (Trichoptera: Hydropsychidae) — J. North Am. Benthol. Soc. 29: 1058-1074. doi:10.1899/09-108.1 ![]()
Peverieri G.S., Simoni S., Goggioli D., Liguori M., Castagnoli M. 2009 — Effects of variety and management practices on mite species diversity in Italian vineyards — Bull. Insect. 62(1): 53-60.
Pons J., Barraclough T.G., Gomez-Zurita J., Cardoso A., Duran D.P., Hazell S., Kamoun S., Sumlin W.D., Vogler A. 2006 — Sequence-based species delimitation for the DNA taxonomy of undescribed insects — Syst. Biol. 55(4): 595-609.
Puillandre N., Modica M.V., Zhang Y., Sirovich L., Boisselier M.-C., Cruaud C., Holford M., Samadi S. 2012b — Large-scale species delimitation method for hyperdiverse groups — Mol. Ecol. 21(11): 2671-91. doi:10.1111/j.1365-294X.2012.05559.x ![]()
Ragusa S., Ciulla A.M. 1991 — Phytoseiid mites associated with vines in Sicilian vineyards — In: The acari. Reproduction, development and life-history strategies (R. Schuster and P.W. Murphy Eds.), Chapman and Hall, pp. 417-423. doi:10.1007/978-94-011-3102-5$_$42 ![]()
Rezende D., Navia D., Mendonça R.S., Melo J.W.S, Gondim M.G.C. 2015 — The predatory mite Neoseiulus paspalivorus (Phytoseiidae) in Brazil: taxonomic status, reproductive compatibility and morphological and molecular variability — Exp. Appl. Acarol. 67: 547-564.
Robinson E.A., Blagoev G.A., Hebert P.D.N., Adamowicz S.J. 2009 — Prospects for using DNA barcoding to identify spiders in species-rich genera — ZooKeys 16: 27-46.
Ronquist F., Huelsenbeck J.P. 2003 — MrBayes 3: Bayesian phylogenetic inference under mixed models — Bioinformatics 19: 1572-1574.
Ros V.I.D., V J.A.J, Menken S.B.J. 2008 — Origins of asexuality in Bryobia mites (Acari: Tetranychidae) — BMC Evolutionary Biology 8: 153. doi:10.1186/1471-2148-8-153 ![]()
Rosenberger M., Maraun M., Scheu S., Schaefer I. 2013 — Pre- and post-glacial diversifications shape genetic complexity of soil-living microarthropod species — Pedobiologia Int. J. Soil Biol. 56 : 79-87.
Rowell H.J., Chant D.A., Hansell R.I.C. 1978 — The determination of setal homologies and setal patterns on the dorsal shield in the family Phytoseiidae (Acarina: Mesostigmata) — Can. Entomol. 110: 859-876.
Roy L., Dowling A.P.G., Chauve C.-M., Buronfosse T. 2010 — Diversity of phylogenetic information according to the locus and the taxonomic level: An example from a parasitic mesostigmatid mite genus — Int. J. Mol. Sci. 11 : 1704-1734. doi:10.3390/ijms11041704 ![]()
Schwarzfeld M.D., Sperling F.A. 2015 — Comparison of five methods for delimitating species in Ophion Fabricius, a diverse genus of parasitoid wasps (Hymenoptera, Ichneumonidae) — Mol. Phylogenet. Evol. 93: 234-248. doi:10.1016/j.ympev.2015.08.003 ![]()
Skoracka, A., Dabert M. 2010 — The cereal rust mite Abacarus hystrix (Acari: Eriophyoidea) is a complex of species: evidence from mitochondrial and nuclear DNA sequences — Bull. Entomol. Res. 100: 263-272. doi:10.1017/S0007485309990216 ![]()
Stalstedt J., Bergsten J., Ronquist F. 2013 — « Forms » of water mites (Acari: Hydrachnidia): intraspecific variation or valid species? — Ecol. Evol. 10: 3415- 3435. doi:10.1002/ece3.704 ![]()
StatSoft France 2010 — STATISTICA (logiciel d'analyse des données), version 9.1. http://www.statsoft.fr.
Sullivan J. 1996 — Combining data with different distributions ofamong-site variation — Syst. Biol. 45: 375-380. doi:10.1093/sysbio/45.3.375 ![]()
Swofford D.L. 2002 — PAUP. Phylogenetic Analysis Using Parsimony and other methods. Ver. 4.0b.10. — Sinauer Associates, Inc., Sunderland, MA, USA.
Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. 2013 — MEGA6: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods — Mol. Biol. Evol. 30: 2725-2729.
Tixier M.-S. 2012 — Approaches to assess intraspecific variations of morphological continuous characters: the case study of the family Phytoseiidae (Acari: Mesostigmata) — Cladistics 28(5): 489-502.
Tixier M.-S. 2013 — Statistical approaches to assess interspecific differences for morphological continuous characters: the case study of the family Phytoseiidae (Acari: Mesostigmata). Zool. Scr. 42(3): 327-334.
Tixier M.-S., Ferrero M., Okassa M., Guichou S., Kreiter S. 2010a — Morphological and molecular analyses of four populations of Phytoseiulus longipes (Mesostigmata: Phytoseiidae) in accordance with their feeding behaviour — Bull. Entomol. Res. 100(5): 569-579. doi:10.1017/S0007485309990617 ![]()
Tixier M.-S., Kreiter S., Barbar Z., Ragusa S., Cheval B. 2006 — The status of two cryptic species: Typhlodromus exhilaratus Ragusa and Typhlodromus phialatus Athias-Henriot (Acari: Phytoseiidae): consequences for taxonomy — Zool. Scr. 35: 115-122. doi:10.1111/j.1463-6409.2006.00222.x ![]()
Tixier M.-S., Kreiter S., Croft B.A., Cheval B. 2008a — Kampimodromus aberrans (Acari: Phytoseiidae) from USA: morphological and molecular assessment of its identity — Bull. Entomol. Res. 98: 125-134.
Tixier M.-S., Kreiter S., Moraes G.J. 2008b — Biogeographic distribution of the mites of the family Phytoseiidae (Acari: Mesostigmata) — Biol. J. Linn. Soc. 93: 845-856.
Tixier M.-S., Okassa M., Liguori M., Poinso A., Salerno B., Kreiter S. 2010b — Voucher specimens for DNA sequences of Phytoseiid mites (Acari: Mesostigmata) — Acarologia 50(4): 487-494. doi:10.1051/acarologia/20101984 ![]()
Tixier M.-S., Okassa M., Kreiter S. 2012 — An integrative morphological and molecular diagnostics for Typhlodromus pyri (Acari: Phytoseiidae) — Zool. Scr. 41: 68-78.
Tixier M.-S., Otto J., Kreiter S, Vicente V., Beard J. 2014 — Is Neoseiulus wearnei the Neoseiulus californicus of Australia? — Exp. Appl. Acarol. 62: 267-277.
Tixier M.-S., Hernandes-Akashi F., Guichou S., Kreiter S. (2011a) — The puzzle of DNA sequences of Phytoseiidae (Acari: Mesostigmata) in the public Genbank database — Inv. Syst. 25: 389-406.
Tixier M.-S., Tsolakis H., Ragusa S., Poinso A., Ferrero M., Okassa M., Kreiter S. 2011b — An integrative taxonomical approach demonstrates the synonymy between Cydnodromus idaeus and C. picanus (Acari: Phytoseiidae) — Inv. Syst. 25: 273-281.
Tsolakis H., Tixier M.-S., Kreiter S., Ragusa S. 2012 — The genus concept within the family Phytoseiidae (Acari: Parasitiformes). Historical review and phylogenetic analyses of the genus Neoseiulus Hughes — Zool. J. Linn. Soc. 165: 253-273.
Valentini A., Pompanon F., Taberlet P. 2009 — DNA barcoding for ecologists — Trend. Ecol. Evol. 24: 110-117. doi:10.1016/j.tree.2008.09.011 ![]()
Wiemers M., Fiedler K. 2007 — Does the DNA barcoding gap exist? A case study in blue butterflies (Lepidoptera: Lycaenidae) — Front. Zool. 4: 1-16.
Wiens J.J. 1998a — Combining data sets with different phylogenetic histories — Syst. Biol. 47: 568-581. doi:10.1080/106351598260581 ![]()
Wiens J.J. 1998b — Does adding characters with missing data increase or decrease phylogenetic accuracy? — Syst. Biol. 47: 625-640. doi:10.1080/106351598260635 ![]()
Wiens J.J., Fetzner J.W., Parkinson C.L., Reeder T.W. 2005 — Hylid frog phylogeny and sampling strategies for speciose clades — Syst. Biol. 54: 719-748. doi:10.1080/10635150500234625 ![]()
Witt J.D.S, Threloff D.L., Hebert P.D.N. 2006 — DNA barcoding reveals extraordinary cryptic diversity in an amphipod genus: implications for desert spring conservation — Mol. Ecol. 15: 3073-3082. doi:10.1111/j.1365-294X.2006.02999.x ![]()
Yang Z., Rannala B. 2012 — Molecular phylogenetics: principles and practice — Nature reviews genetics 13 : 303-314.
Zheng Y., Wiens J.J. 2016 — Combining phylogenomic and supermatrix approaches, and a time-calibrated phylogeny for squamate reptiles (lizards and snakes) based on 52 genes and 4,162 species — Mol. Phylogenet. Evol. 94 : 537-547. doi:10.1016/j.ympev.2015.10.009 ![]()



2016-08-23
Date accepted:
2016-11-30
Date published:
2017-05-05
Edited by:
Faraji, Farid

This work is licensed under a Creative Commons Attribution 4.0 International License
2017 Tixier, Marie-Stéphane; Dos Santos Vicente, Victor; Douin, Martial; Duso, Carlo and Kreiter, Serge
 Download article
Download articleDownload the citation
RIS with abstract
(Zotero, Endnote, Reference Manager, ProCite, RefWorks, Mendeley)
RIS without abstract
BIB
(Zotero, BibTeX)
TXT
(PubMed, Txt)